酶法動態動力學拆分制備R-(-)-乙酰基鄰氯扁桃酸
2014-07-05 16:05:30沈薩薩姜靈陸杰于洪巍
化工進展 2014年9期
沈薩薩,姜靈,陸杰,于洪巍
(1江南大學化學與材料工程學院,江蘇 無錫 214122;2浙江大學化學工程與生物工程學系,浙江 杭州 310028)
酶法動態動力學拆分制備R-(-)-乙酰基鄰氯扁桃酸
沈薩薩1,姜靈2,陸杰1,于洪巍2
(1江南大學化學與材料工程學院,江蘇 無錫 214122;2浙江大學化學工程與生物工程學系,浙江 杭州 310028)
采用假單胞菌脂肪酶Pseudomonassp. ECU1011催化乙酰基鄰氯扁桃酸進行不對稱水解,利用突變后的扁桃酸消旋酶(V29I)對拆分后的產物S-(-)-鄰氯扁桃酸進行消旋,消旋后的鄰氯扁桃酸經過酰化重新被利用到水解反應中,實現了酶法動態動力學拆分制備R-(-)-乙酰基鄰氯扁桃酸。通過對拆分反應、拆分混合物的分離回收以及消旋反應的工藝優化,最終獲得光學純度ee>99.9%的R-(-)-乙酰基鄰氯扁桃酸,其收率達80%。本研究建立的R-(-)-乙酰基鄰氯扁桃酸的動態動力學拆分工藝,對其工業化應用具有重要的指導意義。
酶法動態動力學拆分;脂肪酶Pseudomonassp. ECU1011;扁桃酸消旋酶;乙酰基鄰氯扁桃酸;鄰氯扁桃酸
鄰氯扁桃酸是一種重要的精細化學品,在醫藥化學品領域具有廣泛應用。其中,R-(-)-鄰氯扁桃酸是合成新型血小板聚集抑制劑氯吡格雷的重要中間體[1-3]。隨著氯吡格雷、鄰氯扁桃酸及其衍生物用途的進一步開發,R-(-)-鄰氯扁桃酸的需求量日益增加,但由于受到傳統工藝的限制,市場需求難以得到滿足。
目前,R-(-)-鄰氯扁桃酸的制備主要有以下幾種方法。①直接利用不對稱合成或還原的方法制備光學活性的R-(-)-鄰氯扁桃酸。2009年,Yin等[4]利用金屬釕(Ru)作為催化劑催化不對稱氫化法合成R-(-)-鄰氯扁桃酸甲酯,產物的ee值為92%。但反應條件較苛刻,且有毒的重金屬易污染底物。②非對映體鹽結晶法是目前工業上應用最廣泛的一種拆分技術。它是利用手性試劑將外消旋體混合物中的兩個對映異構體轉化成非對映異構體,然后利用其物理性質的差異將非對映異構體分開,從而得到單一異構體。但其面臨的共同問題是拆分劑價格昂貴,且有一定的毒性,因此在一定的程度上造成了資源的浪費和環境的污染。③酶催化拆分方法是利用具有立體選擇性的酶作為催化劑,在此催化劑的作用下,底物的其中一種異構體優先反應,從而達到把兩種對映異構體轉化成非對映異構體,實現拆分的目的。如報道的商品酶CAL-A(Novozym 735)可以水解拆分鄰氯扁桃酸甲酯,產物ee值高達99%以上[5]。但這種方法的理論收率最多50%,造成了資源的浪費。然而動態動力學拆分技術的出現為解決這個問題提出了新的思路。動態動力學拆分在動力學拆分的基礎上添加了反應活性較弱的對映體的原位消旋,拆分過程與反應活性較弱的對映體原位消旋相繼或同時進行,即某一對映體不斷轉化為產品,而另一對映體則不斷地消旋化補充原料,最終得到理論產率100%的光學純化合物[6-11]。
目前利用動態動力學拆分制備R-(-)-乙酰基鄰氯扁桃酸的研究尚未見報道。如圖1所示,本研究采用假單胞菌脂肪酶Pseudomonassp. ECU1011催化乙酰基鄰氯扁桃酸進行不對稱水解,獲得未反應的R-(-)-乙酰基鄰氯扁桃酸和產物S-(-)-鄰氯扁桃酸[12-13]。R-(-)-乙酰基鄰氯扁桃酸經過水解就可獲得R-(-)-鄰氯扁桃酸。同時,利用扁桃酸消旋酶使鄰氯扁桃酸產物消旋化,形成鄰氯扁桃酸消旋體,經過乙酰化合成拆分底物乙酰基鄰氯扁桃酸,重新作為原料進入反應體系。因此,通過對乙酰基鄰氯扁桃酸進行動態動力學拆分獲得R-(-)-鄰氯扁桃酸前體物是R-(-)-乙酰基鄰氯扁桃酸制備的一種有前景的新工藝。
1 材料與方法
1.1 試劑
Pseudomonassp. ECU1011脂肪酶、乙酰基鄰氯扁桃酸(純度為99.9%)和扁桃酸消旋酶(突變體V29I)由實驗室制備。R-(-)-乙酰基鄰氯扁桃酸和鄰氯扁桃酸(質量分數為99%),由上海精純試劑有限公司購得。氯仿,分析純,購自上海精純試劑有限公司。乙酸乙酯,分析純,購自杭州化工有限公司。其他試劑均為分析純。

圖1 乙酰基鄰氯扁桃酸的動態動力學拆分
1.2 實驗方法
1.2.1 乙酰基鄰氯扁桃酸的拆分反應
配制磷酸緩沖液(200mmol/L,pH=6.2),取10mL置于25mL的錐形瓶中,加入50g/L的脂肪酶Pseudomonassp. ECU1011凍干粉和20mmol/L的底物乙酰基鄰氯扁桃酸,反應體系為密閉體系,將反應體系置于45℃,200r/min的恒溫搖床中,反應5h,獲得R-(-)-乙酰基鄰氯扁桃酸底物和鄰氯扁桃酸產物。
1.2.2S-(-)-鄰氯扁桃酸的消旋反應
配制磷酸緩沖液(200mmol/L,pH=7.5)和濃度10mmol/L的S-(-)-鄰氯扁桃酸。加入10%的鄰氯扁桃酸消旋酶的酶液,封閉反應體系,將反應體系置于30℃,200r/min的恒溫搖床中,反應3.5h,獲得鄰氯扁桃酸的消旋體。
1.2.3 拆分反應后的混合物分離實驗
首先,向拆分反應液中加入不同體積的氯仿萃取溶劑,將該密閉體系置30℃,200r/min的恒溫搖床中,萃取30min。通過萃取收率的大小來確定萃取劑的使用量。然后,根據確定的萃取劑的使用量,分別研究了在20℃、30℃、40℃下,10min、20min、30min、40min、50min時的萃取收率,篩選出最佳萃取溫度和時間。在以上優化條件下,本實驗還研究了萃取次數對萃取效果的影響。每個實驗均設置3個平行樣,取平均值。
1.2.4 乙酰基鄰氯扁桃酸的DKR反應
配制磷酸緩沖液(200mmol/L,pH=6.2),取100mL置于250mL的錐形瓶中,加入50g/L的脂肪酶Pseudomonassp. ECU1011凍干粉和20mmol/L的底物乙酰基鄰氯扁桃酸,將反應體系置于45℃、200r/min的恒溫搖床中,反應5h,獲得R-(-)-乙酰基鄰氯扁桃酸底物和鄰氯扁桃酸產物。拆分反應結束后,將體系中的酶離心去除,調節pH值至7.5,加入10%的鄰氯扁桃酸消旋酶的酶液,將反應體系置于30℃、200r/min的恒溫搖床中,反應3.5h,獲得鄰氯扁桃酸的消旋體。采用溶劑萃取法將未反應的R-(-)-乙酰基鄰氯扁桃酸和鄰氯扁桃酸消旋體分離。將鄰氯扁桃酸的消旋體酰化后得到的乙酰基鄰氯扁桃酸重新用于拆分反應。最終實現動態動力學拆分的整個過程。
1.3 分析方法
分析方法為液相分析法。液相色譜儀為Agilent 1100 series(美國),AD-H柱(4.6nm×250mm),柱溫保持在25℃,流動相為正己烷和異丙醇,流動相配比為90∶10,流速1mL/min,檢測波長λ=254nm,進樣量保持在20~50μL。在此液相條件下,測得S-(+)-乙酰基鄰氯扁桃酸和R-(-)-乙酰基鄰氯扁桃酸、S-(+)-鄰氯扁桃酸和R-(-)-鄰氯扁桃酸的保留時間分別為7.847min、8.450min、13.234min、14.378min。
底物對映體過量值eeS=(CSR-CSS)/(CSR+CSS)× 100%,產物的對映體過量值eeP=(CPS-CPR)/(CPR+CPS)× 100%,轉化率由根據底物的減少計算而得,式中CSS、CSR、CPR和CPS分別為S-(+)-乙酰基鄰氯扁桃酸、R-(-)-乙酰基鄰氯扁桃酸、R-(-)-鄰氯扁桃酸和S-(+)-鄰氯扁桃酸的含量。
2 結果與討論
2.1 乙酰基鄰氯扁桃酸的拆分反應
徐建和課題組[11-12]研究發現假單胞菌脂肪酶Pseudomonassp. ECU1011可以催化乙酰基鄰氯扁桃酸的不對稱水解,得到R-(-)-乙酰基鄰氯扁桃酸和產物鄰氯扁桃酸。此脂肪酶可以優先水解S-(-)-乙酰基鄰氯扁桃酸,并且產物鄰氯扁桃酸的對映體過量值最高可達99%。此反應也可同時得到對映體過量值較高的R-(-)-乙酰基鄰氯扁桃酸。在徐建和課題組研究的基礎上,為了獲得高產率的光學高純度R-(-)-乙酰基鄰氯扁桃酸,對脂肪酶催化拆分反應的條件進行了進一步的優化。
2.1.1 拆分反應的溫度
為了提高R-(-)-乙酰基鄰氯扁桃酸的產率,對拆分反應的溫度進行了進一步的優化研究。如圖2所示,在相同反應時間內,在40℃和45℃反應時R-(-)-乙酰基鄰氯扁桃酸的ee值較高,消旋效果較好。而反應溫度在30℃或55℃時酶活受到溫度的影響,導致R-(-)-乙酰基鄰氯扁桃酸的對映體過量值明顯偏低。因此進一步對40℃和45℃下的R-(-)-乙酰基鄰氯扁桃酸的產率進行了分析。發現由于底物自水解的發生,R-(-)-乙酰基鄰氯扁桃酸的產率隨著反應時間的延長而降低,而在45℃的反應條件下,反應3.5h內(R)-乙酰基鄰氯扁桃酸的產率已經開始降低,此時R-(-)-乙酰基鄰氯扁桃酸對映體過量值還沒有達到理想值。而在溫度為40℃時,自水解比較緩慢,當R-(-)-乙酰基鄰氯扁桃酸的ee>99%時,R-(-)-乙酰基鄰氯扁桃酸的水解量還很少,見圖3。綜合考慮拆分反應后R-(-)-乙酰基鄰氯扁桃酸的對映體過量和產率的最優值,確定了最佳反應溫度為40℃。
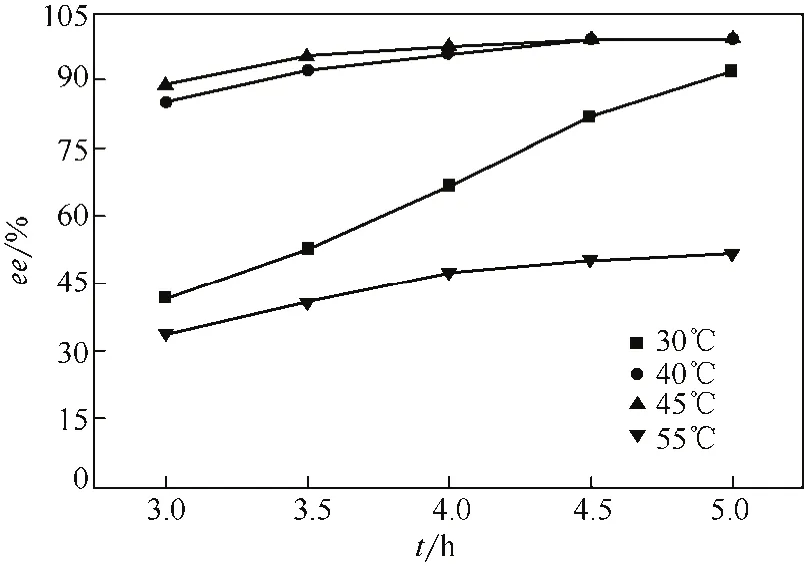
圖2 溫度對R-(-)-乙酰基鄰氯扁桃酸的對映體過量值的影響
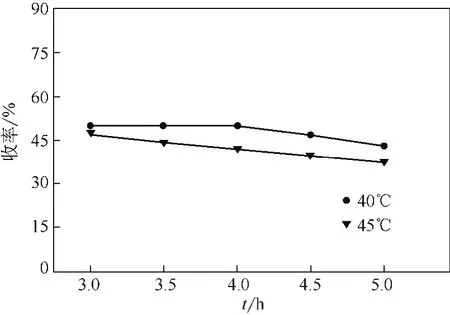
圖3 溫度對R-(-)-乙酰基鄰氯扁桃酸產率的影響

表1R-(-)-乙酰基鄰氯扁桃酸的對映體過量值和產率隨時間的變化
2.1.2 拆分反應時間
拆分反應時間對R-(-)-乙酰基鄰氯扁桃酸的對映體過量值和產率的影響如表1所示。隨著反應時間的增加,(R)-乙酰基鄰氯扁桃酸的對映體過量值也隨之增大,當底物轉化率超過50%時,eeS>99%。但是隨著反應的繼續進行,底物轉化率繼續升高的同時還伴隨著R-(-)-乙酰基鄰氯扁桃酸的量逐漸減少。當反應時間少于4.25h時,未反應的(R)-乙酰基鄰氯扁桃酸的收率基本保持最大收率,但是對映體過量值不高。當反應時間大于4.25h時,未反應的R-(-)-乙酰基鄰氯扁桃酸的收率開始下降,但其對映體過量值穩定在eeS>99.9%。綜合考慮拆分反應后R-(-)-乙酰基鄰氯扁桃酸的對映體過量和收率的最優值,確定最佳反應時間為4.25h。
綜合考慮以上影響拆分效果的因素,從拆分反應中R-(-)-乙酰基鄰氯扁桃酸的對映體過量和收率值的角度,確定了拆分反應的最佳條件:以20mmol/L乙酰基鄰氯扁桃酸作為拆分底物,將反應體系置于40℃、200r/min的恒溫搖床中,反應4.25h。
2.2 鄰氯扁桃酸的消旋反應
消旋酶是一種催化底物產生消旋化的酶,它可以將單一的對映體轉化為消旋體。本實驗中鄰氯扁桃酸的消旋反應就是通過扁桃酸消旋酶催化的。將拆分后產生的鄰氯扁桃酸單一對映體經過扁桃酸消旋酶催化產生消旋體鄰氯扁桃酸。得到的消旋體又可經過乙酰化反應轉化為拆分底物繼續進行拆分反應。從而在理論上可以使R-(-)-乙酰基鄰氯扁桃酸的轉化率提高至100%。因此消旋反應是動態動力學拆分過程中的重要反應。為此本實驗對消旋反應的條件進行了優化,研究了pH值、溫度和鄰氯扁桃酸濃度對消旋反應的影響,確定了消旋反應的最佳反應條件。
2.2.1 溫度對消旋反應的影響
推力桿由推力桿頭、柱管、橡膠襯套總成、彈性擋圈組成,其中橡膠襯套總成由橡膠、銷軸構成。平衡懸架中的推力桿結構如圖2所示。
一般情況下,溫度的變化會使酶的結構發生變化,從而導致選擇性和反應活性發生變化,因此必須考察反應溫度對消旋反應的影響。實驗采用相同反應體系,分別在20℃、25℃、30℃、35℃、40℃的搖床內反應2h。結果如圖4所示:鄰氯扁桃酸的轉化率隨溫度的升高而逐漸增加,當溫度在25℃~30℃時,消旋反應的轉化率達到最高,而隨著溫度的進一步升高,酶活隨著溫度的升高而降低,導致消旋效果進降低,使得消旋反應的轉化率減少。因此消旋反應的最佳溫度范圍為25~30℃。以下實驗溫度均采用30℃。

圖4 溫度對消旋反應的影響
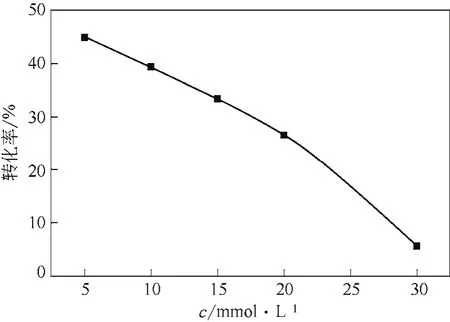
圖5 底物濃度對消旋反應的影響
2.2.2 底物濃度對消旋反應的影響
配制不同濃度的底物S-鄰氯扁桃酸,在相同的反應條件下反應2h。結果如圖5所示:隨著底物濃度的增加,消旋反應的轉化率逐漸減少,消旋效果逐漸降低。原因可能是當底物濃度過高時產生底物抑制效應,對酶促反應的活性產生了消極影響。雖然當底物濃度為5mmol/L時消旋效果最好,可實際得到的產物太少,不利于實驗檢測和回收純化,因此確定10mmol/L為適宜的底物濃度。
2.2.3 pH值對消旋反應的影響
配制不同pH值(6.0~8.0)的緩沖液,在相同反應條件下反應2h,結果如圖6所示:隨著反應體系pH值的增大,消旋反應的轉化率逐漸增加,而當pH=7.5時,反應的轉化率最大,達到39.4%。但隨著pH值的進一步增加,反應的轉化率反而下降。當繼續增加消旋反應的時間(從2h延長到8h),使反應完全進行,結果顯示:當pH值在6~7范圍或大于7.5時,都有一定的消旋效果。在pH=7.5時,消旋反應的轉化率可達50%,底物幾乎完全消旋。因此,消旋反應的最佳pH值為7.5。
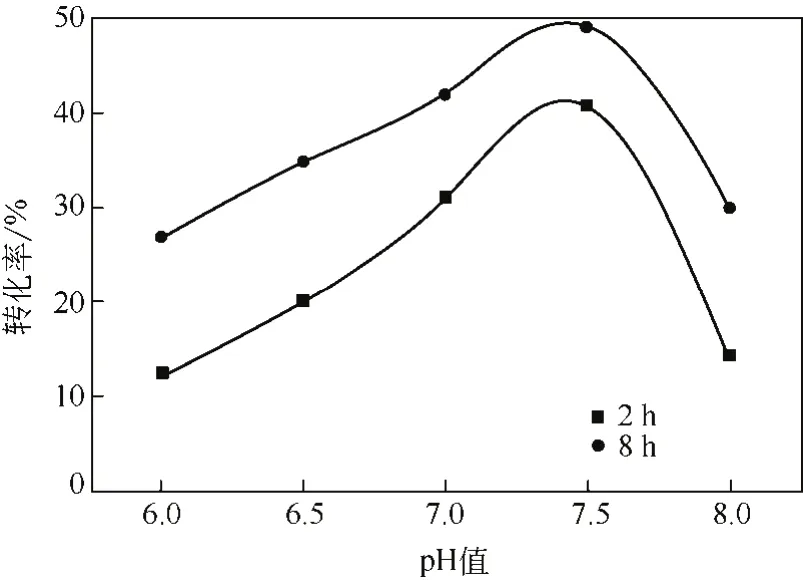
圖6 pH值對消旋反應的影響
2.2.4 反應時間對轉化率的影響
綜合以上的各項因素,采用優化后的條件進行消旋反應,考察了完全反應所需要的時間。結果如圖7所示:隨著反應的進行,消旋反應的轉化率逐漸增加,當反應時間超過3h后,反應的轉化率接近50%,并且趨于平衡狀態。因此反應3h時,消旋反應已經進行完全。
2.3 拆分產物的溶劑萃取分離過程

圖7 pH值7.5時,轉化率隨反應時間的變化
目前對于拆分反應后的混合物主要采用柱層析的方法進行分離。但是此方法分離過程繁瑣,分離量較少,經濟費用較高,不適合工業化應用。工業上較常用結晶和萃取的方法進行分離。從節約成本和降低耗能的角度,采用溶劑萃取法對乙酰基鄰氯扁桃酸拆分后的混合物進行分離。實驗對萃取劑進行了篩選,然后對R-(-)-乙酰基鄰氯扁桃酸萃取劑的使用量、萃取時的溫度和時間、萃取的次數進行了研究。
2.3.1 萃取劑的選擇
本研究從幾種實驗室較常用的有機溶劑中選擇合適的萃取劑,采用乙醚、石油醚、正己烷、氯仿、乙酸乙酯作為萃取劑,分別考察了他們的萃取效果。取相同的酶法制備液,在室溫下,分別加入等體積的上述5種有機溶劑進行1次萃取。萃取結果見表2。結果表明:石油醚和正己烷對R-(-)-乙酰基鄰氯扁桃酸和鄰氯扁桃酸均沒有萃取作用,乙醚和乙酸乙酯對兩種物質都有較高的萃取效果,但這兩種有機溶劑無法達到讓兩者分離的效果。只有氯仿能夠有選擇性的萃取R-(-)-乙酰基鄰氯扁桃酸,而對鄰氯扁桃酸無萃取效果,因此可以實現前者的有效分離。而乙酸乙酯對兩種物質的萃取收率都達到96%以上,因此后續制備余液中的產物鄰氯扁桃酸可直接用乙酸乙酯萃取回收。為達到兩者的有效分離,首先采用氯仿對酶制備液中的R-(-)-乙酰基鄰氯扁桃酸進行萃取分離,再對剩余的余液直接用乙酸乙酯進行萃取,分離回收余液中的鄰氯扁桃酸。
2.3.2 萃取劑用量

表2 不同有機溶劑對R(-)-乙酰基鄰氯扁桃酸和鄰氯扁桃酸的萃取效果
工業中采用有機溶劑進行萃取時,為了減少有機溶劑的用量,減少環境污染,降低成本,會采用最優的萃取劑量進行萃取,以期獲得最大的經濟效益和環境效益。因此對萃取劑氯仿在萃取過程中的使用量進行了研究。實驗考察了氯仿/制備液體積比在(1∶4) ~(2∶1)(即氯仿量為2.5~20mL)時對R-(-)-乙酰基鄰氯扁桃酸萃取效果的影響,見圖8。結果表明,當萃取劑氯仿的體積為10~20mL時,對制備液中R-(-)-乙酰基鄰氯扁桃酸的萃取效果基本保持不變,萃取收率大約在65%。隨著萃取劑體積的增加,萃取收率也不再提高;但萃取劑的體積低于10mL時,由于萃取劑體積比的減小,影響了物質在兩相中的分配,導致萃取收率下降。綜合考慮氯仿對R-(-)-乙酰基鄰氯扁桃酸的萃取回收效果以及過程中的能耗,宜選用等體積的氯仿(10mL)進行萃取,在此條件下的萃取收率為65%。
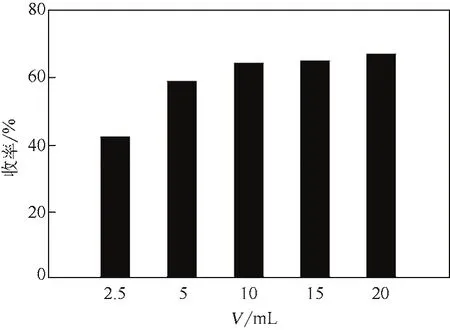
圖8 氯仿用量對R-(-)-乙酰基鄰氯扁桃酸萃取收率的影響
2.3.3 萃取溫度和時間
萃取過程中溫度的改變會導致萃取物的擴散速度以及在兩相分配系數的改變,從而使萃取效果受到較大影響;萃取物在兩相中達到一個分配平衡的狀態需要一定的時間。因此,萃取時需考慮溫度和時間對萃取效果的影響,期望節約成本和能耗,并獲得最好的萃取收率。由于萃取的制備液中有Pseudomonassp. ECU1011脂肪酶的存在,考慮到在萃取過程中水解仍在緩慢進行,為避免R-(-)-乙酰基鄰氯扁桃酸的進一步損失,實驗應避開酶催化反應的最佳溫度(50℃),因此選取20~40℃的溫度范圍進行萃取實驗。溫度以及時間對萃取效果的影響結果見圖9。結果表明:隨著萃取溫度的升高,萃取收率也在提高,而40℃時的萃取收率明顯高于其他兩個溫度下的萃取收率。同時隨著時間的延長,同溫度下的萃取收率逐漸提高。當萃取時間超過40min時,萃取效果趨于穩定。因此在后續的實驗中選擇的萃取條件為:溫度40℃,時間40min。

圖9 萃取溫度和時間對R-(-)-乙酰基鄰氯扁桃酸萃取收率的影響
2.3.4 萃取次數
根據前面確定的萃取條件,氯仿對制備液單次萃取時,R-(-)-乙酰基鄰氯扁桃酸底物的萃取收率僅為75%,為了進一步提高萃取收率,研究考察了萃取次數對底物萃取效果的影響。在40℃的實驗溫度下,采用等體積的氯仿進行萃取,每次萃取時間均為40min。萃取次數對萃取效果的影響結果見圖10。結果表明:在相同的條件下,對酶制備液萃取不同次數后,底物R-(-)-乙酰基鄰氯扁桃酸的萃取收率最高可達到94%。在第2次萃取后,萃取收率明顯升高;而當萃取次數大于2次時,萃取收率變化不大。因此,在后續的實驗中確定適宜的萃取次數為2次。
綜合考慮以上影響萃取效果的各因素,從節約成本和降低耗能的角度,確定R-(-)-乙酰基鄰氯扁桃酸的最優萃取條件為:等體積的氯仿進行萃取,萃取溫度為40℃,萃取次數2次,每次萃取時間均為40min。最終獲得的R-(-)-乙酰基鄰氯扁桃酸的萃取收率為96%。
2.4 產物鄰氯扁桃酸的萃取
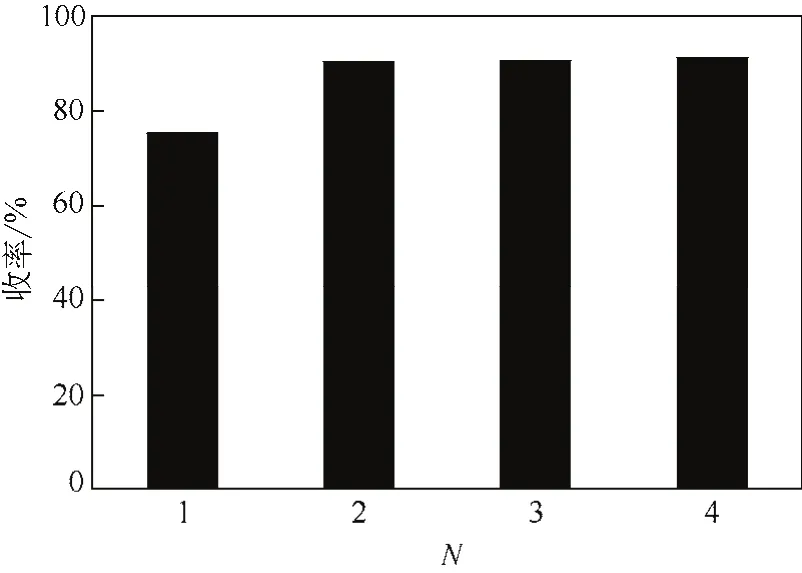
圖10 萃取次數對R-(-)-乙酰基鄰氯扁桃酸萃取收率的影響
萃取劑篩選實驗證明乙酸乙酯對鄰氯扁桃酸具有很好的分離效果(室溫萃取收率達到96%以上),因此采用等體積的乙酸乙酯直接對剩余液進行萃取。考慮一次萃取可能無法達到完美的萃取效果,為了進一步提高產物的萃取收率,將萃取次數提高到2次。最終獲得鄰氯扁桃酸產物的萃取收率為98%。
對溶劑萃取各條件進行優化后,進一步考察了萃取過程對酶反應混合物的實際分離效果。采用乙酸乙酯直接萃取酶法制備液得到的R-(-)-乙酰基鄰氯扁桃酸和鄰氯扁桃酸的混合物[圖11(a)]作為對照,在實驗研究的最優萃取條件下,通過氯仿萃取得到高純度的R-(-)-乙酰基鄰氯扁桃酸底物[圖11(b)],萃取收率為96%。并通過乙酸乙酯萃取剩余液得到高純度的鄰氯扁桃酸產物[圖11(c)],萃取收率為98%。用液相色譜分別對3種萃取物進行檢測。結果表明:采用氯仿和乙酸乙酯依次萃取后,可有效地分離R-(-)-乙酰基鄰氯扁桃酸和鄰氯扁桃酸的混合物,兩者的純度均達到99.9%。

圖11 液相色譜檢測結果
采用以上實驗獲得的優化條件,最終得到R-(-)-乙酰基鄰氯扁桃酸的收率為80%,eeS>99.9%。
3 結 論
采用動態動力學方法對乙酰基鄰氯扁桃酸消旋體進行拆分制備R-(-)-乙酰基鄰氯扁桃酸。為獲得高產率光學純的R-(-)-乙酰基鄰氯扁桃酸,實驗對拆分反應和消旋反應過程分別進行了條件優化。在此優化條件下,乙酰基鄰氯扁桃酸消旋體拆分獲得R-(-)-乙酰基鄰氯扁桃酸的產率為47%,對映體過量值ee>99%。將拆分后的混合物通過氯仿和乙酸乙酯進行萃取分離后,將反應得到的鄰氯扁桃酸進行消旋化,經過3h后就可獲得完全消旋化的鄰氯扁桃酸消旋體。將消旋體鄰氯扁桃酸轉化成乙酰基扁桃酸消旋體原料后,繼續作為原料用于拆分反應。反應循環4次,最終得到的R-(-)-乙酰基鄰氯扁桃酸的收率為80%。此試驗研究打破了傳統動力學拆分反應產物產率低的局限,實現了高產率光學純R-(-)-乙酰基鄰氯扁桃酸的制備分離過程。
[1] 唐田,王彥青,王海全. 氯吡格雷硫酸氫鹽的合成[J]. 中國醫藥工業雜志,2009,40(50):324-325.
[2] Abbert D,Ferran C,Mafrand J P. Thieno[3,2-c] pyridine derivatives and their therapeutical use:EP,0099802[P]. 1987-04-02.
[3] 陳子明,杜玉民,鮑春和. 氯毗格雷合成路線圖[J]. 中國醫藥工業雜志,2002,33(4):206-208.
[4] Yin L,Shan W J,Jia X. Ru-catalyzed enantioselective preparation of methyl (R)-o-chloromandelate and its application in the synthesis of (S)-clopidogrel[J].Journal of Organometallic Chemistry,2009,694:2092-2095.
[5] Uhm K N,Lee Y H. Enantioselective resolution of methyl 2-chloromandelate byCandida antarcticalipase A in a solvent-free transesterification reaction[J].Journal of Molecular Catalysis B:Enzymatic,2007,45:34-38.
[6] 張占輝,劉慶彬. 酶過渡金屬配合物催化的動態動力學拆分研究進展[J]. 有機化學,2005,25(7):780-787.
[7] 馬紅敏,邵瑞鏈,成俊然,等. 動力學拆分法的研究進展[J]. 有機化學,2000,4(20):454-463.
[8] Héléne P. Recent developments in dynamic kinetic resolution[J].Tetrahedron,2008,64:1563-1601.
[9] 張占金,毛金城,萬伯順,等. 路易斯酸堿催化的外消旋體(動態)動力學拆分反應[J]. 化學進展,2004,16(4):574-583.
[10] Schnell B,Faber K,Kroutil W. Enzymatic racemisation and its application to synthetic biotransformations[J].Advanced Synthesis & Catalysis Adv.,2003,345:653-666.
[11] 杜志強,王安明,王華. 手性化合物的動態動力學拆分研究進展[J].分子催化,2008,22(5):1001-3555.
[12] Xin J,Jiang P,Yu H L,et al. ImprovingPseudomonassp. esterase performance by engineering approaches for kinetic resolution of 2-acetoxyphenylacetic acids[J].Biochemical Engineering Journal,2011,57:63-68.
[13] Xin J,Jiang P,Yu H L,et al. Bioproduction of chiral mandelate by enantioselective deacylation ofα-acetoxyphenylacetic acid using whole cells of newly isolatedPseudomonassp. ECU1011 [J].Applied Microbiology and Biotechnology,2010,86:83-91.
R-(-)-acetyl-o-mandelic acid preparation via enzymatic dynamic kinetic resolution
SHEN Sasa1,JIANG Ling2,LU Jie1,YU Hongwei2
(1School of Chemical and Material Engineering,Jiangnan University,Wuxi 214122,Jiangsu,China;2School of Chemical and Biological Engineering,Zhejiang University,Hangzhou 310028,Zhejiang,China)
In this study,asymmetric hydrolysis of acetyl chloromandelic acid was catalyzed byPseudomonassp. ECU1011,and theS-(-)-chloromandelic acid in the resolution mixture was then racemerized using the mutated mandelate racemase and fed back into the hydrolysis reaction after acylation,realizing enzymatic dynamic kinetic resolution for preparation ofR-(-)-acetyl chloromandelic acid. Firstly,the conditions for enzymatic resolution,separation of resolution mixture,and enzymatic racemization were optimized. Subsequently,dynamic kinetic resolution of acetyl chloromandelic acid was conducted under the optimal conditions,obtaining optically pureR-(-)-acetyl chloromandelic acid with final yield of 80%. The conditions for enzymatic resolution,chloromandelic acid recycling and racemization were optimized in this study,leading to preliminary establishment of the dynamic kinetic resolution process forR-(-)-acetyl chloromandelic acid preparation,with guiding significance for future industrial applications.
enzymatic dynamic kinetic resolution;Pseudomonassp. ECU1011 lipase;mandelate racemase;R-(-)-acetyl-o-mandelic acid;chloromandelic acid
TQ 463;Q 819
A
1000-6613(2014)09-2425-07
10.3969/j.issn.1000-6613.2014.09.032
2014-01-02;修改稿日期:2014-01-22。
國家自然科學基金(21176102、21176215、GZ935)、江蘇省自然科學基金(BK20131100)、江蘇省環保科研課題基金(2012004)及浙江省自然科學基金杰出青年基金(21076181)項目。
沈薩薩(1985—),女,碩士。E-mail sasashen@163.com。聯系人:陸杰,男,教授,從事結晶分離和藥物合成等研究。E-mail lujie@jiangnan.edu.cn。
