三維有序結構In摻雜TiO2薄膜的可見光催化活性
2014-09-17 06:59:40王景聲王恩君于彥龍郭麗梅曹亞安
物理化學學報 2014年3期
王景聲 王恩君 于彥龍 郭麗梅 曹亞安,*
(1南開大學物理科學學院,天津300071;2南開大學泰達應用物理研究院,弱光非線性光子學教育部重點實驗室,天津300457;3中國科學院合肥物質科學研究院,合肥230031)
1 引言
TiO2具有光催化效率高和化學穩定性強等特點,多年來在光催化研究領域倍受重視.1-3然而,TiO2的禁帶寬度是3.2 eV,僅吸收紫外光(λ>387.5 nm),而對可見光無吸收(太陽光譜中紫外光部分僅占8%,而可見光占45%以上),從而導致了太陽能利用率的降低.另外,TiO2光生載流子的復合導致了光催化效率的降低.
金屬或非金屬摻雜是提高TiO2催化劑可見光催化活性的有效方法.Wang等4制備了Fe摻雜的TiO2催化劑,其光催化活性高于純TiO2.Jing、5Fresno6和Yu7等對Sn摻雜TiO2進行了研究,均發現其光催化活性有明顯提高.此外,TiO2摻雜La3+、Pd2+、Cr3+、Ag+以及稀土金屬離子(Sm3+、Nd3+、Pr3+)等也見報道.8-12近年來Cao等報道了Sn、13Zr、14Ni、15In、16N17和B18等離子摻雜TiO2,結果表明,金屬或非金屬離子摻雜增強了TiO2的可見光響應,促進了光生載流子的分離,有效地提高了催化劑的可見光催化活性.
三維有序結構TiO2薄膜具有比表面積大、吸附能力強和光的利用率高等特點,表現出優異的光催化活性,近年來已成為光催化領域研究的熱點.19-22King等23采用低溫原子層沉積的方法制備了反蛋白石結構TiO2;Ren等24用模板法制備了具有光子禁帶的反蛋白石結構的TiO2,紫外光催化結果表明,該催化劑光子效率比P25高248%;Doong等25制備了高度有序的反蛋白石結構的TiO2,其孔徑范圍在480-1000 nm之間,結果表明,該催化劑的紫外光催化的降解率隨著孔徑的增大而線性提高;Li和Shang26制備了N摻雜反蛋白石結構TiO2,研究發現該催化劑在可見光區吸收增強、光催化活性明顯提高.
直至目前,三維有序結構In離子摻雜的TiO2薄膜可見光催化劑未見報道;對于In離子的摻雜方式和摻雜能級等摻雜機理尚不清楚;In離子摻雜對催化劑光生載流子過程和光催化活性的影響,以及可見光催化機理還有待于研究.
本文采用自組裝生長聚苯乙烯膠體模板方法和溶膠-凝膠法制備出三維有序結構In摻雜TiO2薄膜可見光催化劑.與TiO2和三維有序結構TiO2薄膜相比,該催化劑的可見光催化活性顯著提高.利用X射線電子衍射(XRD)譜、X射線光電子能譜(XPS)和紫外-可見漫反射吸收光譜(UV-Vis DRS)等表征技術,確定了催化劑的結構、In離子的摻雜方式和能帶結構,并分析討論了三維有序結構In摻雜TiO2薄膜降解甲醛的可見光催化機理.
2 實驗部分
2.1 樣品的制備
將洗凈的載玻片垂直放入盛有聚苯乙烯微球的懸濁液中(懸濁液的體積為8.5 mL,聚苯乙烯微球的體積分數為0.2%),在干燥箱中55°C恒溫沉化,待溶劑完全蒸發后,載玻片表面生長出聚苯乙烯微球膠體薄膜,然后在80°C加熱2 h,得到聚苯乙烯微球膠體模板.
室溫下將 4.4 mL 的 InCl3溶液(0.6 mol·L-1)與40 mL無水乙醇混合,在劇烈攪拌下,將12 mL的Ti(OC4H9)4緩慢滴加到上述混合液中,然后再加入一定量的鹽酸(12 mol·L-1),調節pH為3.5,經劇烈攪拌后,得到穩定透明的TiO2溶膠.
將聚苯乙烯微球膠體模板垂直浸泡在TiO2溶膠中(約1 min),以2.5 mm·s-1的速率垂直提拉成膜,空氣中自然干燥,上述過程重復兩次.將模板移至馬弗爐中,以5°C·min-1的速率緩慢升溫至500°C,焙燒2 h后自然冷卻,制備出三維有序結構In摻雜的TiO2(IO-TiO2-In)薄膜,放入干燥器備用.
三維有序結構TiO2(IO-TiO2)薄膜制備方法同上,區別為TiO2溶膠中未加入InCl3.采用垂直提拉法制備出TiO2溶膠薄膜,500°C焙燒2 h(升溫速率為5 °C·min-1),制得純TiO2薄膜.
實驗中所用試劑均為分析純,實驗用水為ρ≥18.0 MΩ·cm高純水.
2.2 物性表征
X射線衍射(XRD,Rigaku D/max-2500,Cu靶,Kα線)測定了薄膜樣品的晶體結構;X射線光電子能譜(XPS)的測試在ESCA Lab 250型X光電子能譜儀(Mg-Al靶,Kα線)上進行,所有的譜圖依據C 1s(284.6 eV)進行校正;利用掃描電子顯微鏡(SEM,JEOL JSM-6700F)對薄膜樣品的表面形貌和厚度進行觀測;利用紫外-可見分光光度計(U-4100,Hitachi)測定薄膜樣品的紫外可見漫反射吸收光譜.
2.3 光催化反應
光催化降解甲醛的實驗在密封的Pyrex玻璃反應器(455 mL)中進行,以500 W的Xe燈作為催化反應的外照光源,輻射波長>290 nm(在可見光催化實驗中,反應器前放置400 nm的截止濾光片);薄膜催化劑平鋪在反應器底部(面積為2 cm×4 cm),膜面朝光,并與光照方向保持垂直,光源距反應器10 cm;反應器內加入15 μL甲醛(優級純99.9%),在干燥箱中60°C恒溫40 min,使甲醛充分揮發形成氣體;反應器與光源之間放置隔熱玻璃,恒溫保持反應體系的溫度((30±2)°C)與濃度(1.19×10-3mol·L-1)的平衡.利用氣相色譜儀(GC-7890F,天美)每隔80 min(紫外光40 min)監測甲醛光催化降解生成CO2的濃度變化;色譜柱為TDX-01(1 m×? 3 mm),高純氮氣(99.99%)為載氣,在氫火焰檢測器與色譜柱間裝有轉化爐,使CO2發生加氫反應(CO2+4H2→CH4+2H2O),從而測定CO2的濃度.
3 結果與討論
3.1 光催化活性
采用光催化降解甲醛生成CO2(最終產物)的濃度來評價催化劑的光催化活性.圖1為TiO2、IO-TiO2和IO-TiO2-In薄膜可見光催化降解甲醛生成CO2的曲線,其可見光催化活性見表1.結果表明,在可見光光解實驗中,光照160 min僅有微量的CO2產生(2.99×10-9mol·L-1);與光解實驗相比,TiO2薄膜幾乎無可見光活性(CO2濃度為2.99×10-9mol·L-1);IOTiO2薄膜的可見光活性(CO2濃度為4.01×10-9mol·L-1,CO2生成率為 8.55×10-11mol·cm-2·h-1)高于TiO2;然而,IO-TiO2-In薄膜表現出最高可見光活性(CO2濃度為1.89×10-8mol·L-1,CO2生成率為4.03×10-10mol·cm-2·h-1),其產生 CO2濃度和 CO2生成率大約是TiO2和IO-TiO2的5倍.圖2為TiO2、IO-TiO2和IO-TiO2-In薄膜紫外光催化降解甲醛生成CO2的曲線,其紫外光催化活性見表1.樣品的紫外光催化活性遵循IO-TiO2-In>IO-TiO2>TiO2規律,與可見光催化相似.
3.2 催化劑的性質
圖3為TiO2、IO-TiO2和IO-TiO2-In薄膜的SEM照片.結果表明,TiO2薄膜(圖3a)是由堆積致密的TiO2納米粒子組成,其粒徑約為20-40 nm.IO-TiO2薄膜(圖3b)呈現三維周期有序的多層多孔結構,每層由六邊形骨架連接而成,相鄰六邊形中心間距約為200 nm.IO-TiO2-In薄膜(圖3c)的結構與IO-TiO2相同.
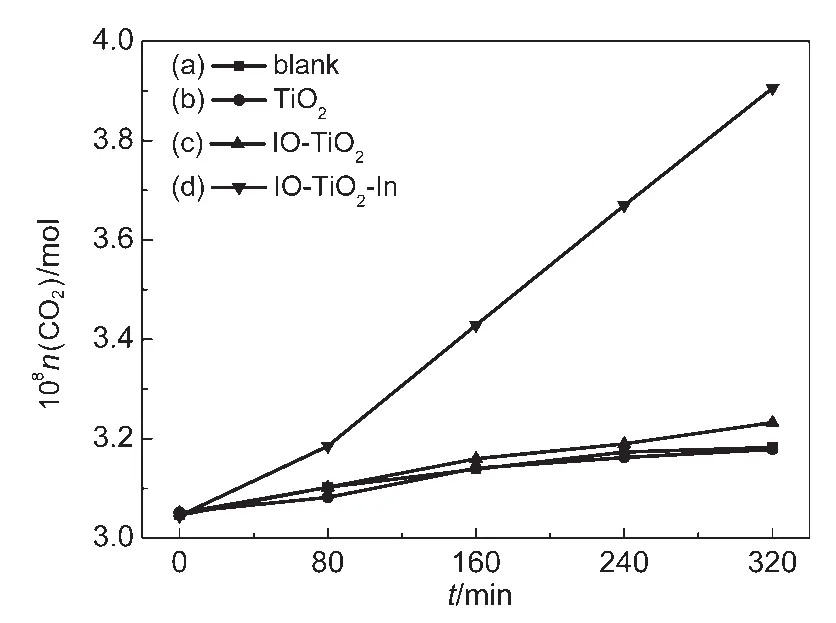
圖1 TiO2(b)、IO-TiO2(c)和IO-TiO2-In(d)薄膜可見光降解甲醛生成CO2的曲線Fig.1 CO2generation curves of photocatalytic degradation of formaldehyde under visible light with TiO2(b),IO-TiO2(c),and IO-TiO2-In(d)films(a)Blank is the photolysis of HCHO.

圖2 TiO2(b)、IO-TiO2(c)和IO-TiO2-In(d)薄膜紫外光降解甲醛生成CO2的曲線Fig.2 CO2generation curves of photocatalytic degradation of formaldehyde under UV light with TiO2(b),IO-TiO2(c),and IO-TiO2-In(d)films(a)Blank is the photolysis of HCHO.
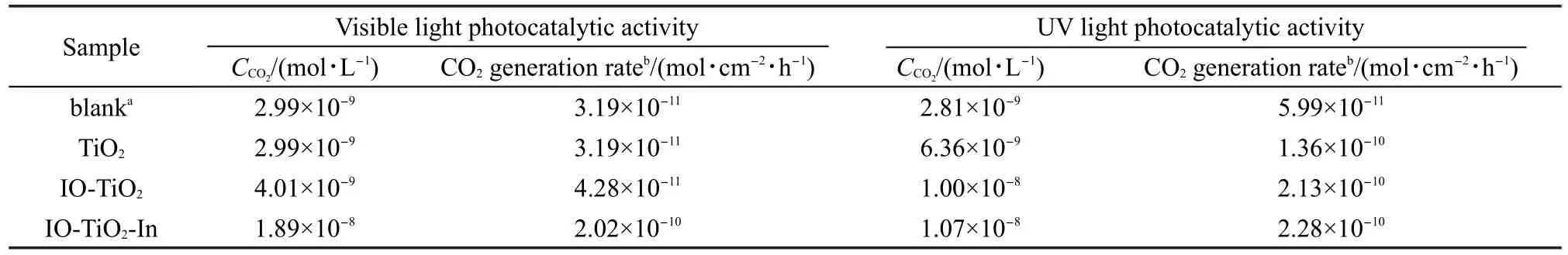
表1 TiO2、IO-TiO2和IO-TiO2-In薄膜可見和紫外光催化活性Table 1 Photocatalytic activity of TiO2,IO-TiO2,and IO-TiO2-In films under visible and UV light irradiation

圖3 TiO2(a)、IO-TiO2(b)和IO-TiO2-In(c)薄膜的掃描電子顯微鏡(SEM)照片Fig.3 Scanning electron microscopy(SEM)images of TiO2(a),IO-TiO2(b),and IO-TiO2-In(c)films
圖4 為TiO2、IO-TiO2和IO-TiO2-In薄膜的XRD譜.在25.2°、37.8°、48.0°、53.8°和 54.9°附近的衍射峰分別對應于銳鈦礦 TiO2(101)、(004)、(200)、(105)和(211)晶面,27表明所有樣品主要為四方晶系銳鈦礦結構.在圖4(c)中,22.3°、30.9°、45.5°和58.2°附近出現的衍射峰符合In2O3的衍射峰(ICSD 21-1272),表明在摻雜過程中In離子在TiO2表面可能形成In2O3物種.以上結果能夠由XPS譜進一步證明.圖5為TiO2、IO-TiO2和 IO-TiO2-In薄膜的Ti 2p的 XPS譜.結合能為458.4和464.2 eV的峰分別為銳鈦礦TiO2的晶格 Ti的 Ti 2p3/2和 Ti 2p1/2;16而在 TiO2、IOTiO2和IO-TiO2-In薄膜的O 1s的XPS譜(圖6)中,結合能為529.6、530.7和532.0 eV的峰(XPSPEAK41軟件擬合)分別歸屬為銳鈦礦TiO2的晶格氧、表面橋氧和表面羥基(OH)氧,28由此證明TiO2、IO-TiO2和IO-TiO2-In薄膜樣品均為銳鈦礦結構.,
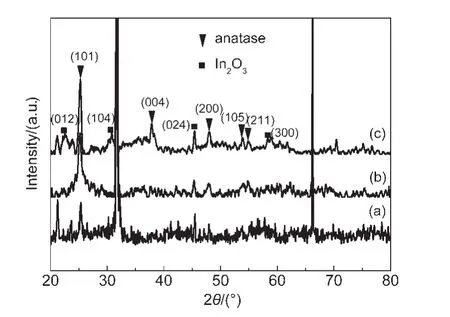
圖4 TiO2(a)、IO-TiO2(b)和IO-TiO2-In(c)薄膜的X射線衍射(XRD)譜Fig.4 X-ray diffraction(XRD)spectra of TiO2(a),IO-TiO2(b),and IO-TiO2-In(c)films
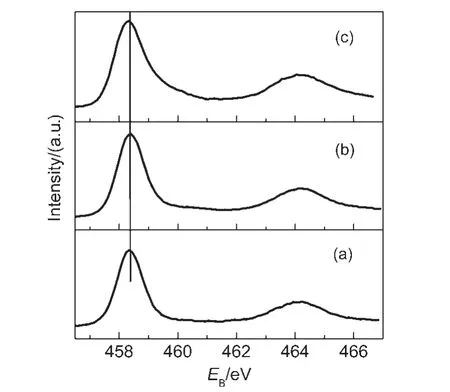
圖5 TiO2(a)、IO-TiO2(b)和IO-TiO2-In(c)薄膜的Ti 2p的X射線光電子能譜(XPS)圖Fig.5 Ti 2p X-ray photoelectron spectroscopies(XPS)for TiO2(a),IO-TiO2(b),and IO-TiO2-In(c)films
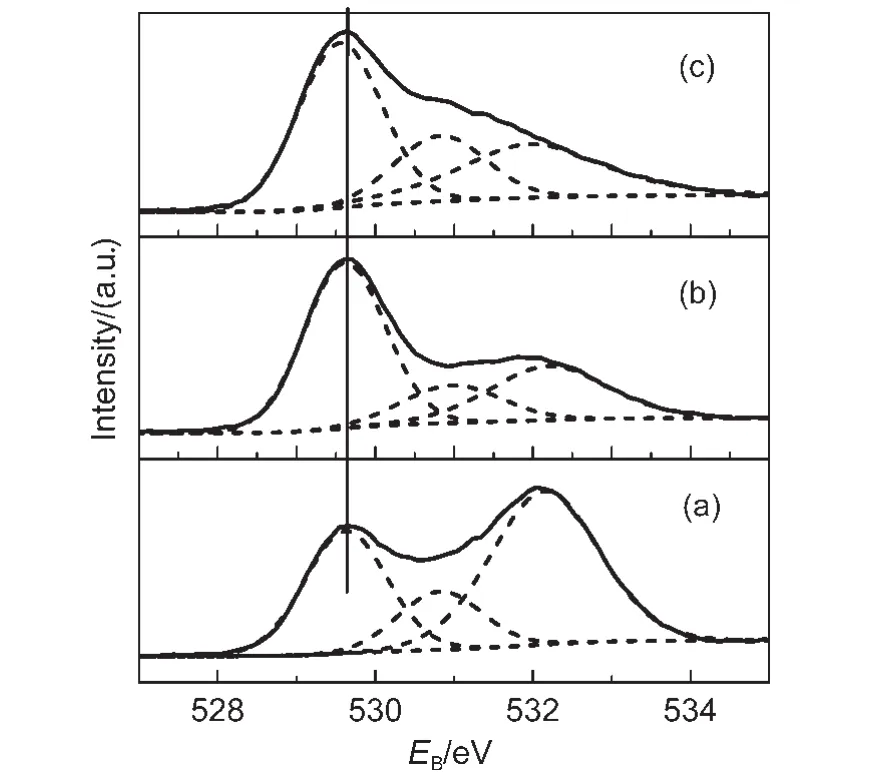
圖6 TiO2(a)、IO-TiO2(b)和IO-TiO2-In(c)薄膜的O 1s的XPS圖Fig.6 O 1s XPS for TiO2(a),IO-TiO2(b),and IO-TiO2-In(c)films
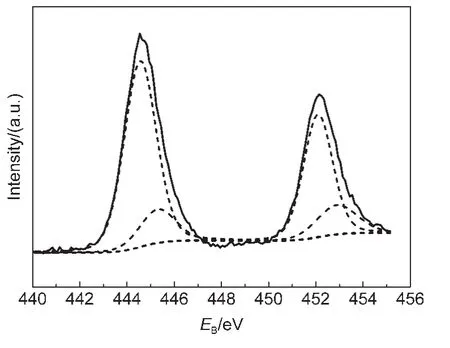
圖7 IO-TiO2-In薄膜In 3d的XPS譜Fig.7 In 3d XPS for IO-TiO2-In films
圖7 為IO-TiO2-In薄膜In 3d的XPS譜.擬合后,在結合能為444.6和452.1 eV的一對強峰與In2O3中In 3d5/2和In 3d3/2的結合能一致,29表明摻入的In離子主要是以In2O3物種的形式存在于IO-TiO2-In薄膜表面,這與XRD表征結果一致.另外,基于XPS原理分析,由于In的電負性(1.7)大于Ti的電負性(1.5),如果In離子進入TiO2的晶格,取代并占據Ti離子的晶格位置,將會導致TiO2中晶格Ti和晶格O的結合能提高.16然而,與TiO2比較,IO-TiO2-In薄膜的Ti 2p和O 1s譜(圖5和圖6)中的晶格Ti 2p和晶格O 1s的峰未發生明顯高位移動,進一步表明摻雜的In離子沒有進入TiO2晶格,而是作為In2O3物種存在于TiO2表面,形成TiO2與In2O3復合結構(TiO2/In2O3).
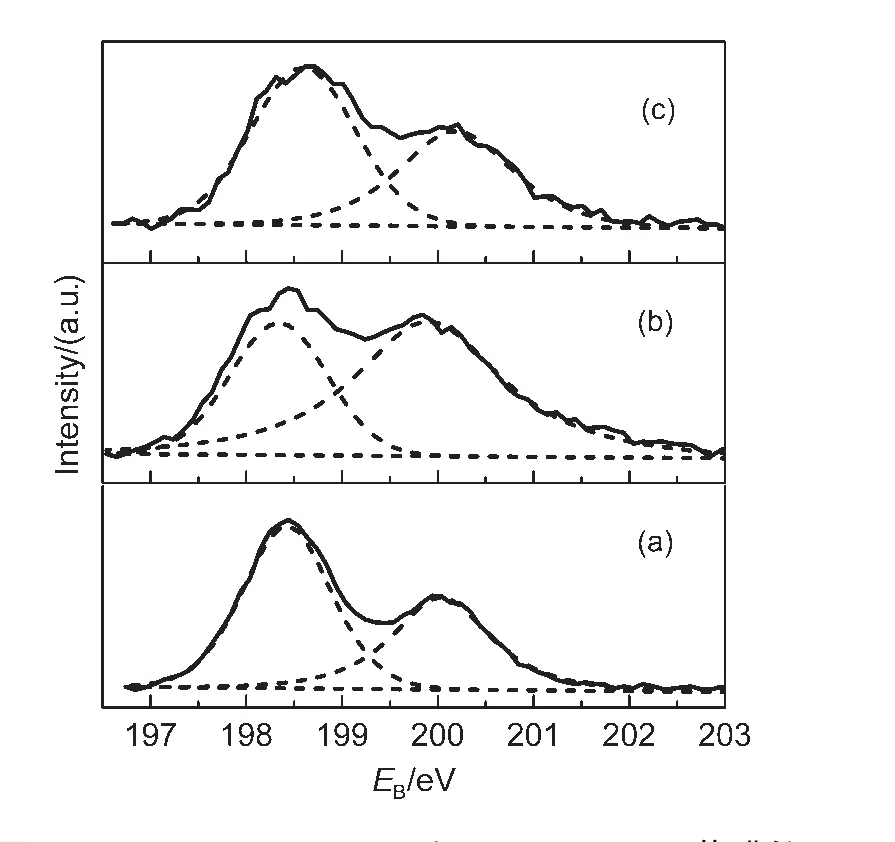
圖8 TiO2(a)、IO-TiO2(b)和IO-TiO2-In(c)薄膜的Cl 2p的XPS圖Fig.8 Cl 2p XPS for TiO2(a),IO-TiO2(b),and IO-TiO2-In(c)films
然而,在擬合后的In 3d譜中,445.3和452.9 eV的一對弱峰,其In 3d5/2的結合能(445.3 eV)位于In2O3中In 3d5/2的結合能(444.6 eV)30和 InCl3中 In 3d5/2的結合能(446.0 eV)31之間,意味著摻入的In離子可能與O和Cl離子連接.對于IO-TiO2-In薄膜Cl 2p3/2譜(圖8c),Cl 2p3/2的結合能(198.64 eV)高于TiCl4中Cl 2p3/2的結合能(198.20 eV),而低于InCl3中Cl 2p3/2的結合能(199.1 eV),31表明Cl離子可能與In連接.然而Cl的離子半徑(0.181 nm)大于O離子半徑(0.140 nm),不可能進入TiO2晶格取代晶格O,Cl僅能存在于樣品表面.32基于以上分析,在IO-TiO2-In薄膜中In離子的摻雜態能夠被確定:In離子分別與表面的O和Cl離子連接,在IO-TiO2-In薄膜表面形成O-In-Clx(x=1,2)物種.對于TiO2和IO-TiO2薄膜(圖8(a,b)),Cl 2p3/2峰的結合能分別為198.36和198.39 eV,與TiCl4的Cl 2p3/2結合能(198.2 eV)33接近,表明 Cl?離子能夠與Ti4+連接,在TiO2和IO-TiO2表面形成Ti-Clx(x=1,2)物種.
圖9為TiO2、IO-TiO2和IO-TiO2-In薄膜的紫外-可見光漫反射吸收譜.在小于400 nm的強峰為TiO2的帶-帶躍遷.起峰閾值分別為374.7、374.9和382.5 nm,相應的禁帶寬度為3.37、3.33和3.12 eV.另外,IO-TiO2-In樣品在375-480 nm出現的肩峰為表面In2O3物種的價帶到導帶的躍遷(In2O3的禁帶寬度為2.8 eV34);而500-800 nm吸收峰歸屬為價帶到O-In-Clx物種的表面態能級的躍遷,表面態能級約在導帶下0.5-1.6 eV范圍.16以上結果表明,In離子的摻雜在IO-TiO2-In薄膜的表面形成In2O3和O-In-Clx物種,既增強了IO-TiO2-In薄膜的可見光吸收,又有利于可見光催化活性的提高.
3.3 光催化機理
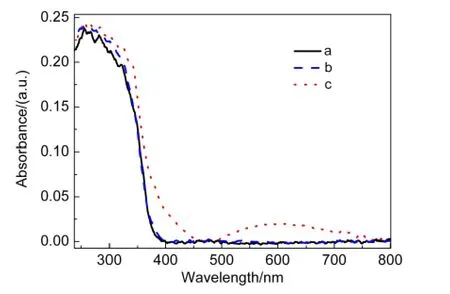
圖9 TiO2(a)、IO-TiO2(b)和IO-TiO2-In(c)薄膜的紫外-可見光漫反射吸收光譜Fig.9 UV-Vis diffuse reflectance spectra of TiO2(a),IO-TiO2(b),and IO-TiO2-In(c)films
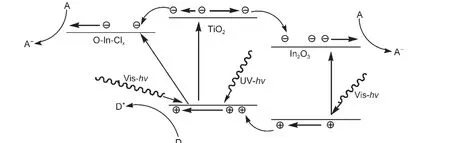
圖10 IO-TiO2-In薄膜的能帶結構及可見和紫外光催化機理示意圖Fig.10 Schematic diagram of energy band structures and photocatalytic mechanism under visible and UV light for IO-TiO2-In film
依據以上表征結果,IO-TiO2-In薄膜的能帶結構見圖10,其可見、紫外光催化機理如下.對于可見光催化,TiO2和IO-TiO2薄膜可見光活性低的主要原因是TiO2和IO-TiO2的禁帶寬度(3.20 eV)較大,對于可見光(λ>400 nm)沒有吸收.與TiO2和IO-TiO2薄膜相比,IO-TiO2-In薄膜具有較高的可見光活性主要歸因于In離子摻雜.In離子摻雜在IO-TiO2-In薄膜表面主要形成In2O3和O-In-Clx(x=1,2)物種;In2O3的禁帶寬度為2.8 eV,34可以吸收可見光,提高了可見光利用率;而TiO2和In2O3的復合有效地提高了光生載流子的分離(阻止其復合).35另外,O-In-Clx(x=1,2)物種的表面態能級在導帶下0.5-1.6 eV范圍,在可見光(λ>400 nm)的照射下,催化劑產生價帶到表面態能級的躍遷,增加了可見光的吸收,導致了光生載流子的直接分離.由In2O3的帶-帶躍遷和TiO2導帶到O-In-Clx表面態能級躍遷產生的光生電子能夠被表面吸附的O2分子俘獲形成O-2,進一步氧化表面吸附甲醛分子.In2O3價帶的光生空穴經TiO2/In2O3界面,與TiO2價帶的光生空穴一起直接氧化表面吸附甲醛分子,導致了IO-TiO2-In薄膜催化劑可見光催化活性的有效提高.
對于紫外光催化,IO-TiO2-In薄膜相對TiO2和IO-TiO2薄膜紫外光活性進一步提高的原因主要決定于催化劑的能帶結構(圖10).TiO2和IO-TiO2薄膜具有較高的紫外光活性歸因于帶-帶躍遷的光生電子和光生空穴在催化劑表面的氧化還原作用;36然而,導帶大部分的光生電子在轉移到表面過程中由無輻射躍遷到氧空位能級與價帶的光生空穴復合,僅有少數的光生電子和光生空穴到達催化劑的表面參加表面甲醛分子降解反應.對于IO-TiO2-In薄膜,TiO2帶-帶躍遷的光生電子可以轉移到O-In-Clx(x=1,2)物種的表面態能級或轉移In2O3導帶到達催化劑表面;In2O3價帶的光生空穴能夠轉移到TiO2價帶,與TiO2價帶的光生空穴一道到達催化劑的表面.與TiO2和IO-TiO2薄膜相比,以上電荷轉移過程有效地提高了光生載流子的分離效率,阻止光生載流子復合,能夠使更多的光生電子和光生空穴到達催化劑的表面參加表面甲醛分子降解反應,導致了催化劑紫外光催化活性的有效提高.
另外,IO-TiO2和IO-TiO2-In薄膜具有三維有序結構,與TiO2薄膜相比,比表面積大,光的利用率高,更有利于紫外和可見光催化活性的提高.
4 結論
采用自組裝生長聚苯乙烯膠體模板法和溶膠-凝膠法制備出高活性的三維有序結構In摻雜TiO2薄膜可見光催化劑(IO-TiO2-In).與TiO2相比,IOTiO2-In薄膜不但比表面積大,光的利用率高,而且表面形成In2O3和O-In-Clx(x=1,2)物種,即增強了可見光的吸收,又促進了光生載流子的分離,有效地提高了光生電子和光生空穴在固/氣界面參加光催化反應的利用率,使IO-TiO2-In薄膜催化劑的可見光催化活性顯著提高.
(1)Choi,W.;Termin,A.;Hoffmann,M.R.J.Phys.Chem.1994,98,13669.doi:10.1021/j100102a038
(2) Ghicov,A.;Macak,J.M.;Tsuchiya,H.;Kunze,J.;Haeublein,V.;Frey,L.;Schmuki,P.Nano Lett.2006,6,1080.doi:10.1021/nl0600979
(3)Chen,X.B.;Mao,S.S.Chem.Rev.2007,107,2891.doi:10.1021/cr0500535
(4)Wang,C.;Bahnemanna,D.W.;Dohrmannb,J.K.Chem.Commun.2000,1539.
(5) Jing,L.Q.;Fu,H.G.;Wang,B.Q.;Wang,D.J.;Xin,B.F.;Li,S.D.;Sun,J.Z.Appl.Catal.B 2006,62,282.doi:10.1016/j.apcatb.2005.08.012
(6) Fresno,F.;Tudela,D.;Coronado,J.M.;Fernández-Gracía,M.;Hungría,A.B.;Soria,J.Phys.Chem.Chem.Phys.2006,8,2421.doi:10.1039/b601920j
(7)Yu,J.G.;Liu,S.W.;Zhou,M.H.J.Phys.Chem.C 2008,112,2050.doi:10.1021/jp0770007
(8) Huo,Y.N.;Zhu,J.;Li,J.X.;Li,G.S.;Li,H.X.Journal of Molecular Catalysis A:Chemical 2007,278,237.doi:10.1016/j.molcata.2007.07.054
(9) Wu,C.;Chao,C.;Kuo,F.Catal.Today 2004,97,103.doi:10.1016/j.cattod.2004.04.055
(10) Anpo,M.;Takeuchi,M.J.Catal.2003,216,505.doi:10.1016/S0021-9517(02)00104-5
(11)Wang,P.;Wang,D.J.;Xie,T.F.;Li,H.Y.;Yang,M.;Wei,X.Mater.Chem.Phys.2008,109,181.doi:10.1016/j.matchemphys.2007.11.019
(12) Liang,C.H.;Li,F.B.;Liu,C.S.;Lu,J.L.;Wang,X.G.Dyes and Pigments 2008,76,477.doi:10.1016/j.dyepig.2006.10.006
(13)Cao,Y.Q.;He,T.;Zhao,L.S.;Wang,E.J.;Yang,W.S.;Cao,Y.A.J.Phys.Chem.C 2009,113,18121.doi:10.1021/jp9069288
(14)Wang,E.J.;Yang,H.Y.;Cao,Y.A.J.Chem.2009,67,2759.
(15) Luo,D.C.;Zhang,L.L.;Long,H.J.;Chen,Y.M.;Cao,Y.A.Acta Phys.-Chim.Sin.2008,24,1095.[羅大超,張蘭蘭,龍繪錦,陳詠梅,曹亞安.物理化學學報,2008,24,1095.]doi:10.3866/PKU.WHXB20080632
(16)Wang,E.J.;Yang,W.S.;Cao,Y.A.J.Phys.Chem.C 2009,113,20912.doi:10.1021/jp9041793
(17) Cao,Y.Q.;He,T.;Chen,Y.M.J.Phys.Chem.C 2010,114,3627.doi:10.1021/jp100786x
(18)Yuan,J.X.;Wang,E.J.;Chen,Y.M.;Yang,W.S.;Yao,J.H.;Cao,Y.A.Appl.Surf.Sci.2011,257,7335.doi:10.1016/j.apsusc.2011.03.139
(19) Chen,J.I.L.;Freymann,G.;Choi,S.Y.;Kitaev,V.G.;Ozin,A.Adv.Mater.2006,18,1915.
(20) Chen,I.L.;Freymann,G.V.;Kitaev,V.;Ozin,G.A.J.Am.Chem.Soc.2007,129,1196.doi:10.1021/ja066102s
(21) Chen,J.I.L.;Loso,E.;Ebrahim,N.;Ozin,G.A.J.Am.Chem.Soc.2008,130,5420.doi:10.1021/ja800288f
(22) Chen,J.I.L.;Freymann,G.;Choi,S.Y.;Kitaev,V.;Ozin,G.A.J.Mater.Chem.2008,18,369.doi:10.1039/b708474a
(23) King,J.S.;Graugnard,E.;Summers,C.J.Adv.Mater.2005,17,1010.
(24) Ren,M.;Ravikrishna,R.;Valsaraj,K.T.Environ.Sci.Technol.2006,40,7029.doi:10.1021/es061045o
(25) Doong,R.A.;Chang,S.M.;Hung,Y.C.Sep.Purif.Technol.2007,58,192.doi:10.1016/j.seppur.2007.07.029
(26) Li,Q.;Shang,J.K.J.Am.Chem.Soc.2008,91,660.
(27) Gao,B.F.;Ma,Y.;Cao,Y.A.;Yang,W.S.;Yao,J.N.J.Phys.Chem.B 2006,110,14391.doi:10.1021/jp0624606
(28)Cao,Y.A.;Yang,W.S.;Chen,Y.M.;Du,H.;Yue,P.Appl.Surf.Sci.2004,236,223.doi:10.1016/j.apsusc.2004.04.020
(29) Li,J.;Zeng,H.C.J.Am.Chem.Soc.2007,129,5839.
(30) Reddya,B.M.;Chowdhury,B.;Smirniotis,P.G.Appl.Catal.A 2001,219,53.doi:10.1016/S0926-860X(01)00658-5
(31) Freeland,B.H.;Habeeb,J.J.;Tuck,D.G.Can.J.Chem.1977,55,1527.doi:10.1139/v77-213
(32) Zhu,J.;Zheng,W.;He,B.;Zhang,J.L.;Anpob,M.J.Mol.Catal.A:Chem.2004,216,35.doi:10.1016/j.molcata.2004.01.008
(33) Mousty-Desbuquoit,C.;Riga,J.;Verbist,J.J.J.Chem.Phys.1983,79,26.doi:10.1063/1.445567
(34) Poznyak,S.K.;Talapin,D.V.;Kulak,A.I.J.Phys.Chem.B 2001,105,4816.doi:10.1021/jp003247r
(35) Cao,Y.A.;Zhang,X.T.;Yang,W.S.;Du,H.;Bai,Y.B.;Li,T.J.;Yao,J.N.Chem.Mater.2000,12,3445.doi:10.1021/cm0004432
(36) Long,H.J.;Wang,E.J.;Dong,J.Z.;Wang,L.L.;Cao,Y.Q.;Yang,W.S.;Cao,Y.A.J.Chem.2009,67,1533.
猜你喜歡
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
現代企業(2015年9期)2015-02-28 18:56:50
