無菌配制系統所存的問題及其保障措施
2014-10-15 09:14:50申志峰
機電信息 2014年23期
申志峰
(湖南千山制藥機械股份有限公司,湖南長沙410081)
0 引言
藥液的配制就是按工藝規程要求把各活性成分、輔料以及溶解成分進行配制,并按順序進行混合,制成批配制溶液,以待下一步的灌裝。配制可以包括固體活性成分的溶解,或者簡單的液體混合,還可以包括更為復雜的操作,例如乳化或者脂質體的形成[1]。
無菌藥品是指法定藥品標準中列有無菌檢查項目的制劑和原料藥,包括無菌制劑和無菌原料藥[2]。無菌藥品從字面上理解首先應該是無菌的,其次應該符合藥品安全、有效、均一、穩定的特性,在生產過程中應最大限度降低微生物、各種微粒和熱原的污染。無菌藥品按生產工藝可分為2類:采用最終滅菌工藝的為最終滅菌產品;部分或全部工序采用無菌生產工藝的為非最終滅菌產品[2]。無菌藥品中的滴眼劑、小容量注射劑、大容量注射劑、凍干粉針劑在生產過程中都需要經過配制工序,在配制工序的稱量投料過程,原輔料會暴露在操作環境中,可以說配制工序是無菌藥品過程質量控制的關鍵,它是直接決定無菌藥品質量尤其是非最終滅菌產品質量的關鍵工序之一。
1 配制工序的污染來源
在配制工序中,原輔料會有一段時間暴露在生產操作環境中,它是引發微生物污染和細菌內毒素污染風險相對較大的環節。無菌藥品在配制工序可能會受到的污染來源有下列幾種:物料帶來的污染、人員操作帶來的污染、配制工藝設計不合理帶來的污染和配制系統帶來的污染等。
1.1 物料帶來的污染
原輔料一般儲存在非潔凈的倉儲區,由于物流程序設計不合理,在轉移過程中可能將污染物帶入潔凈區而引起污染,或者原輔料包裝的破損會直接導致原輔料污染。
未對原輔料的微生物和細菌內毒素建立控制標準和方法,這些沒有經過微生物和細菌內毒素含量檢驗的原輔料,其質量無疑處于不可控制狀態,使用這些原輔料將極大地增加無菌藥品生產的質量風險。
在無菌工藝中使用非無菌的原輔料生產非最終滅菌產品,這些不純正的原輔料直接帶入大量的雜質、不溶性顆粒、微生物和細菌內毒素,降低了無菌工藝的可靠性,導致產品質量很難得到保障。
稱量用的工器具沒有進行滅菌處理,在原輔料接觸過程中給原輔料接種上細菌,增加藥液除菌過濾前的微生物負荷,間接提高了產品的細菌內毒素含量。
1.2 人員操作帶來的污染
人員是無菌藥品生產中主要的污染源,有統計顯示無菌藥品生產過程超過70%的污染來源于人[3]。在藥液配制過程,一方面,通過人員對操作環境的污染間接影響產品帶菌量;另一方面,在某些生產操作中,操作人員與原輔料或藥液可能相互接觸,從而直接污染產品。
1.3 配制工藝設計不合理帶來的污染
目前,液體無菌制劑配制工藝有濃配-稀配兩步法和一步法2種[1]。由于使用的原料含有大量雜質,采用濃配-稀配兩步法一般會在濃配工序加入活性炭以吸附分子量較大的雜質。活性炭能吸附雜質也能引起污染,一是活性炭中的可溶性雜質進入藥液后無法祛除,污染藥液;二是活性炭顆粒極細,容易飛入污染潔凈區和凈化空調系統,引起整個生產系統的污染。加入的活性炭需在濃配工序進行循環脫碳,循環脫碳管線不易清洗和消毒,容易引起污染和交叉污染。在無菌工藝生產中采用濃配-稀配兩步法生產非最終滅菌產品具有較大的質量風險。
1.4 配制系統帶來的污染
在無菌工藝中,配制系統直接決定了每一批產品的質量。由配制系統自身帶來的污染主要有:配制罐上傳動部件的磨損落下的不溶性微粒、潤滑和密封油脂的泄漏或滲出物[4]、系統在線清洗不徹底的殘留物、在線滅菌不徹底遺留的微生物、系統滅菌后發生倒灌引起的污染等。
2 常見配制系統所存的問題
配制系統一般由不同功能的罐體、攪拌系統、控制閥門、儀表、工藝管線、過濾器、計量系統等組成,它是無菌生產工藝的起點,直接決定著藥液的質量和產品的安全,有時也是引起污染和交叉污染的主要途徑。在無菌生產工藝中,配制系統如果在攪拌裝置、計量方式、藥液輸送方式、過濾器使用等方面的使用方式不科學,常常容易引起污染和交叉污染,導致系統潛在風險增加。
2.1 攪拌裝置
攪拌裝置起到將原輔料在分散介質中進行分散混合的作用,目前配制系統常見的方式為上攪拌。上攪拌裝置通過軸將攪拌槳和傳動電機連接起來,軸與罐體連接采用機械密封,密封機構通常填充石墨作為潤滑劑。機械密封在長期使用過程中由于熱脹冷縮會出現密封不嚴的現象,導致潤滑劑滲出污染藥液,增加藥品質量風險,而且這個過程很難通過肉眼觀察到。
上攪拌裝置的傳動軸通常是偏心安裝的,因為罐頂中央通常為注射用水的入口,以方便清洗時注射用水噴灑均勻,但是如果使用上攪拌裝置,攪拌軸會遮擋一部分噴灑出來的注射用水,在罐壁形成一塊注射用水無法到達的扇面(圖1),導致這部分罐壁無法清洗,這不符合在線清洗的要求,而且濺出的水流也會影響周圍水流的沖擊力。采用上攪拌裝置,配制間需增加高度以方便上攪拌裝置發生故障時維修,因為上攪拌裝置維修可能需要將攪拌軸拆卸出來,這就需要配制間有足夠高的空間能夠取出攪拌軸,這樣會增加系統長期運行的費用,并且房間高度太高也不方便清潔和潔凈環境的控制。
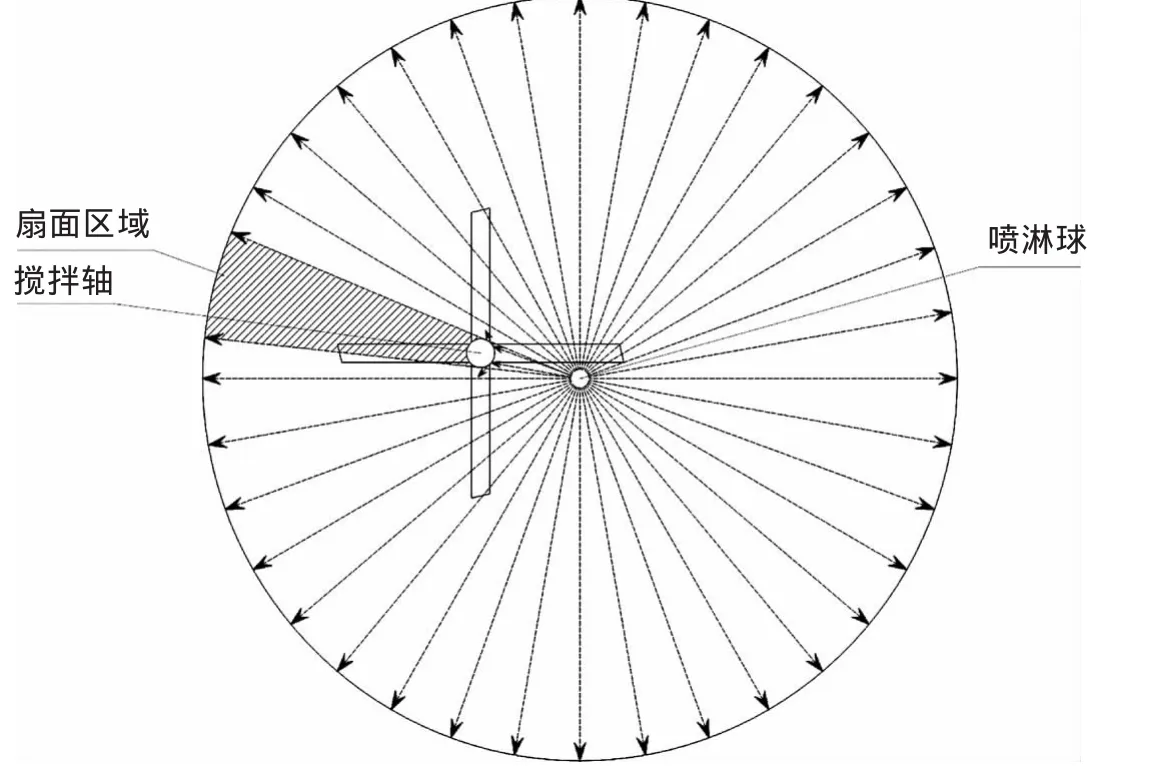
圖1 上攪拌裝置不能清洗區域示意
2.2 計量方式
藥液配制過程需要按工藝規程,在不同的工藝步驟對藥液進行準確計量,傳統的配制系統通常采用液位計的方式計量。液位計有玻璃管液位計和新型電磁感應式液位計,通過液面的高低判斷罐內藥液的容積,它計量精確度不高,通常與藥液有直接接觸,對制造材料也有較高的要求,而這點在藥廠通常會被忽略。玻璃管液位計安裝在罐體外面,與配液罐形成連通器以方便觀察罐內液面的變化,但是液位計內壁無法噴淋,即在配液罐使用完成后,玻璃管液位計無法進行在線清洗。新型電磁感應式液位計安裝在配液罐內部,但是清洗時也會形成無法清洗的扇面。隨著制藥工藝和設備的改進,在無菌工藝設備上應避免使用液位計。
2.3 藥液輸送方式
由于工藝需要,配制完畢混合均勻的藥液需要輸送到下一工序或轉移到無菌容器儲存,通常我們會用泵作為藥液輸送的動力。采用泵輸送藥液,泵前端為正壓,泵后端為負壓,泵后管線泄漏會將環境中的空氣吸入污染藥液。如果流量前后不一致,還需增加回流管線來平衡輸入和輸出的量,例如把藥液輸送給灌裝機。系統使用完畢回流管線需要循環沖洗,不能夠形成淋洗[4]效果,會影響系統在線清洗的效果。采用泵輸送藥液,由離心泵結構和工作原理可知,藥液輸送殘留量較大,而且在滅菌后泵體會殘留較多的水,影響產品的質量。由于回流管線將藥液輸送管線上的過濾器前后聯通,導致系統沒有明確的無菌界限。如果生產人員操作不當,葉輪可能會發生氣蝕現象,而侵蝕下來的金屬微粒進入藥液,將這樣的系統用于無菌藥品生產會引起很高的產品質量風險。
3 無菌配制系統的保障措施
采用無菌生產工藝生產非最終滅菌產品,在生產全過程都應防止各種污染的發生,配制工序決定了一批產品的初始質量特性,沒有一套先進的無菌配制系統就難以保證藥液的質量和產品的安全。無菌配制系統必須具備在線清洗和在線滅菌功能,以保證系統清洗和滅菌效果的重現性,它在設計、安裝和零部件的使用上應盡量消除系統內的各種殘留,降低系統使用的污染風險。
3.1 攪拌裝置
無菌配制系統的攪拌裝置應選擇下置磁力攪拌器。下置磁力攪拌器通過磁力傳動,安裝有攪拌電機和攪拌槳的罐底板與罐壁采用焊接的方式連接,攪拌電機和磁力攪拌槳完全隔離,沒有潤滑劑滲漏的風險,也不用擔心因熱脹冷縮而引起滅菌蒸汽泄漏的問題。由于下置磁力攪拌器安裝在配液罐的底部,清洗時不會因為攪拌軸阻擋噴淋的注射用水而影響罐壁的沖洗,形成無法清洗的扇面區域。攪拌槳一般采用鏤空設計,無死角方便系統的清潔,如圖2所示。采用下置磁力攪拌器應選擇耐高溫的攪拌器,尤其是攪拌器的陰陽軸承應能承受高溫(135℃以上),以適應在線滅菌和工藝加熱的要求,陰陽軸承材質應為化學惰性,無溶出物,不催化藥品分解,同時應該經過驗證確保其在運行過程中不產生脫落物。

圖2 磁力攪拌器內部液流示意圖
3.2 計量方式
無菌配制系統的計量方式應首選稱重模塊在線稱量的方式。稱重模塊安裝在配液罐的支腿上,不會對藥液產生任何影響,也不影響配液罐的清洗和滅菌。稱重模塊能夠迅速反應罐內重量的變化,計量準確可靠,可以有效地防止漏液跑料事故的發生,完全避免了使用液位計的諸多缺陷。但是,由于傳統工藝中,配制工序都是按體積計算的,使用稱重模塊計量需要對生產工藝進行相應變更,以適應硬件的需要。
3.3 藥液輸送方式
無菌配制系統應采用無菌壓縮氣體作為藥液輸送的動力來源,這樣可以有效避免因使用泵作為藥液輸送動力時系統內藥液的殘留,解決系統清洗滅菌不徹底和回流管線無法滅菌等一系列問題。采用無菌壓縮氣體作為藥液輸送動力,藥液在系統內密閉正壓運行,產品不會因系統密封等問題受到污染,最大限度地降低質量風險和生產成本。無菌壓縮氣體應該是無菌、無油、無水、無塵的,但目前還沒有一個統一且嚴格的無菌壓縮氣體標準,只能參考《GB/T 13277—2008壓縮氣體》和《ISO 8573—2001壓縮氣體》制定相應的企業標準和檢測方法。
3.4 其他
無菌配制系統對閥門的選型和安裝、工藝管線的布局、施工和安裝、在線滅菌溫度監測點的設置等都有特殊的要求。例如,公用介質分配閥門應優先使用T型閥(圖3),以最大限度地降低盲管長度。

圖3 T型閥內部結構示意圖
無菌配制系統上采取的所有措施,其最終的目的都是為了最大限度地降低系統的殘留和使用的質量風險,確保患者的用藥安全。
4 無菌配制系統的確認和驗證
確認和驗證是制藥企業基本的質量活動,并且已經成為法規要求。確認與驗證的范圍和定義有所不同:確認主要針對設備、人員和供應商,而驗證則是將經過確認的人員、設備、物料、軟件、程序等整合在一起,證明整個工藝或方法能夠達到既定目的[5]。無菌配制系統的確認和驗證也應符合這一要求。
4.1 確認
用戶需求標準(簡稱URS)在驗證中處于最為基礎的地位,也是進行其他驗證的準繩[6],無菌配制系統的URS編寫應結合公司生產工藝的要求、國家相關行業標準法規要求、公用設備設施的要求、公司對系統安裝施工、文件、服務和培訓等的要求提出具體而詳細的標準。設計確認是用戶與系統的供應商協商確定無菌配制系統的設計符合用戶需求標準。
無菌配制系統的安裝確認包括:收集查驗無菌配制系統相關技術文件并歸檔;檢查施工人員資質和相關設備設施并形成文件記錄;檢查無菌配制系統各部件的安裝施工;關鍵部件的材質材料確認和表面光潔度確認;壓力測試、管線清洗和鈍化確認等活動。
無菌配制系統的運行確認包括按照批準的運行確認方案確認系統各項運行參數符合要求。
無菌配制系統的性能確定,即結合配制系統清潔效果驗證、滅菌效果驗證、培養基灌裝模擬試驗和生產工藝驗證檢查配制系統的各項性能。
4.2 驗證
無菌配制系統的驗證包括配制系統清潔效果驗證、滅菌效果驗證、培養基灌裝模擬試驗和生產工藝驗證。通常用無菌生產工藝生產非最終滅菌產品,藥液配制過濾后需要保存在無菌儲罐內無菌存放一段時間直至產品灌裝完畢,企業應該驗證產品的最長無菌存放時間。國際制藥工程組織(ISPE)在《制藥工程基準指南系列——無菌生產設施》中指出,在某些情況下,有必要將除菌過濾后的料液保存在貯罐里,保持無菌狀態,然后從貯罐為灌裝機供料。如果產品以這樣的方式儲存,應當驗證產品在最長的儲存時間內仍保持無菌[7]。
總之,無菌配制系統的確認和驗證是藥品生產企業驗證體系的一部分,它也沒有一個嚴格的劃分,確認和驗證的范圍和程度應根據風險評估的結果確認[7]。
5 配制工藝的選擇
目前國內無菌藥品生產配制工藝有濃配-稀配兩步法和一步配制法2種工藝。采用濃配-稀配兩步法工藝通常是由于使用的原輔料不純,原輔料中夾帶不溶性微粒、熱原、微生物,這些雜質在制劑工廠很難通過有效的手段除去,目前企業普遍只是簡單地在濃配過程中添加活性炭對溶液中的內毒素進行吸附,而且活性炭的吸附效果也沒有經過驗證。配制過程中添加活性炭通過循環脫碳去除,活性炭可以吸附分子量較大的雜質,如細菌內毒素,但是活性炭中的可溶性雜質進入藥液后不易除去,還會污染潔凈區和凈化空調系統[1],引起污染和交叉污染。
目前,工業發達國家已普遍淘汰了加活性炭濃配-過濾-稀配的傳統工藝,而采用不加活性炭的一步配制工藝。一步配制法避免了傳統工藝的風險[1]。采用一步配制法的前提是有穩定可靠的無菌原料供應。《藥品生產質量管理規范(2010年修訂)附錄1:無菌藥品》第52條指出,應當盡可能減少物料的微生物污染程度。必要時,物料的質量標準中應當包括微生物限度、細菌內毒素或熱原檢查項目[2]。采用無菌工藝生產非最終滅菌產品,應制定嚴格的原輔料的微生物限度、細菌內毒素控制指標,以降低生產過程的微生物和細菌內毒素的荷載,降低產品質量風險,目前普遍采用的100 CFU/g的微生物控制指標是不可接受的。
綜上所述,在無菌工藝中使用無菌配制系統生產非最終滅菌產品應該采用一步配制工藝,這樣更有利于產品的質量控制。
6 結語
基于質量源于設計的理念,在無菌藥品生產的開始就采用先進的工藝和設備,是無菌工藝成功的關鍵和產品質量的有力保障。很多企業往往因重視終端灌裝設備忽略配制系統而導致無菌工藝失敗,例如采用先進的吹、灌、封三合一設備灌裝,而配制系統卻不能夠實現在線清洗和滅菌,同時使用質量不符合要求的原輔料,在灌裝開始前細菌內毒素就已經超標,怎么能生產出合格的產品呢?目前已經出現一次性使用配液系統,無需清潔和滅菌驗證,小批量配液使用靈活方便,還有新型活性炭過濾器和超濾工藝也進入配制應用領域,這些新技術、新工藝將促使配制系統的持續革新。
無菌工藝作為產品質量最難控制的制藥工藝之一,在工藝設計和設備選型上應考慮整個工藝的完整性,這樣才能降低風險,保證生產藥品的質量安全。
[1]國家食品藥品監督管理局藥品認證管理中心.藥品GMP指南(無菌藥品)[M].中國醫藥科技出版社,2011
[2]中華人民共和國衛生部.藥品生產質量管理規范(2010年修訂)[S]
[3]陳西勇,于淑渤,梁宏.無菌藥品生產發生微生物污染的因素分析(上)[J].首都醫藥,2009(3)
[4]周立法.藥液配制系統的無菌質量保證措施[J].機電信息,2012(35)
[5]劉禹.制藥設備URS、IQ、OQ和PQ的組織和連接[J].醫藥工程設計,2008,29(3)
[6]田耀華.制藥機械URS的概念與要素[J].機電信息,2008(17)
[7]ISPE.制藥工程基準指南系列——無菌生產設施[S]
猜你喜歡
中國合理用藥探索(2022年1期)2022-11-26 00:22:32
山東冶金(2019年6期)2020-01-06 07:45:54
世界農藥(2019年2期)2019-07-13 05:55:12
當代陜西(2019年7期)2019-04-25 00:22:18
領導決策信息(2018年26期)2018-10-12 02:18:26
中國衛生(2016年5期)2016-11-12 13:25:28
銅業工程(2015年4期)2015-12-29 02:48:39
中國衛生(2015年5期)2015-11-08 12:09:48
都市麗人(2015年5期)2015-03-20 13:33:49
中國衛生(2014年7期)2014-11-10 02:33:02
