俄羅斯卡爾梅克共和國發酵蔬菜中細菌多樣性研究*
2014-12-16 08:04:32侯強川郭壯張家超孫天松
食品與發酵工業 2014年7期
侯強川,郭壯 ,張家超,孫天松
(內蒙古農業大學乳品生物技術與工程教育部重點實驗室,內蒙古呼和浩特,010018)
發酵蔬菜是以蔬菜為原料,通過自然發酵或人工接種益生菌發酵的方式得到的一大類食品,其產品主要包括泡菜、酸菜、醬菜、腌菜等。發酵蔬菜不僅保留了蔬菜原來的營養價值,同時在有益微生物的作用下,其還能產生多種人體必須的氨基酸和脂肪酸,增加了蔬菜的營養價值,延長了蔬菜的保質期。
既往研究表明,乳酸菌對于發酵蔬菜的發酵成熟起著至關重要的作用[1-3],分離篩選品質優良的乳酸菌菌株是發酵蔬菜工業化生產所必不可少的。除此之外,發酵蔬菜中微生物的種類和數量對發酵蔬菜的風味和質量具有極其重要的影響[4]。全面深入地分析發酵蔬菜中微生物的多樣性和菌群組成對于掌握發酵蔬菜風味物質的產生和變化規律,營養物質的生成機理以及科學地預測產品保質期具有重要的意義。
傳統檢測發酵蔬菜中微生物組成最常用的是基于純培養的方法,但受培養條件的限制,該方法并不能完全揭示產品中微生物菌群的組成。之前有研究表明,自然界中只有不到1%的微生物可培養[5],這表明單純以純培養的方式研究發酵蔬菜中微生物的組成,結果往往是低估了產品中微生物的多樣性。伴隨分子生物學的發展,越來越多的基于非培養的微生物檢測方法和技術如DGGE/TGGE技術、克隆文庫分析法、SSCP技術、基因芯片技術等被廣泛應用于發酵蔬菜微生物檢測中。這些新技術的使用使人們對于自然界中99%以上不可培養微生物的研究成為可能。隨著羅氏454公司新一代高通量焦磷酸測序儀的面世,我們獲得了一種全新認識微生物世界的途徑,454焦磷酸測序技術具有測序通量大、快速、成本低等優點,近年來該技術在各類食品微生物檢測中的應用越來越廣泛,如奶酪[6-7],開菲爾[8-9],酒精飲料[10]等發酵食品。
本研究以采集自俄羅斯卡爾梅克共和國埃利斯塔市郊的2份發酵蔬菜為研究對象,通過純培養的方法結合16S rRNA測序技術對其中分離出的乳酸菌進行了準確的鑒定和分型,同時采用454焦磷酸測序技術分析了樣品中細菌構成的多樣性。以期為發酵蔬菜工業化生產菌株的篩選做前期菌株儲備,同時也為全面掌握發酵蔬菜中細菌的組成和改進發酵蔬菜的制作工藝提供有益的參考。
1 材料與方法
1.1 材料與試劑
本實驗的2份發酵蔬菜樣品Ru12、Ru13采集自俄羅斯埃利斯塔市郊,MRS液體培養基、MRS固體培養基、10%脫脂乳培養基等參照文獻[11-13]制備,M17固體培養基(CM0785B)和M17液體培養基(CM0817B)由英國OXOID公司提供,PCR引物由上海桑尼生物科技有限公司合成。Taq酶(DR001C)由寶生物工程(大連)有限公司提供。
1.2 儀器與設備
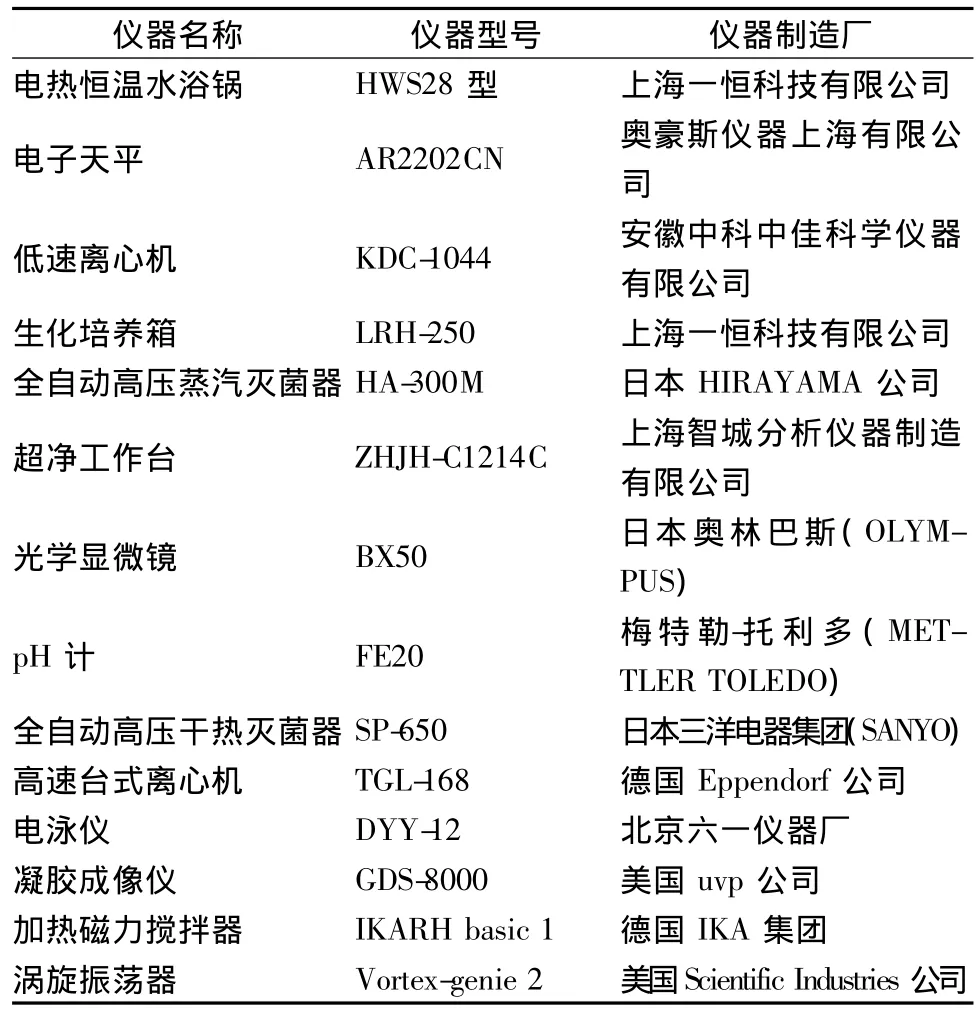
表1 實驗所用的主要儀器Table 1 The main equipment in this study
1.3 菌株培養分離及鑒定方法
1.3.1 乳酸菌的分離純化
無菌條件下,用滅菌槍頭吸取500 μL發酵液于4.5 mL的生理鹽水(0.85%,w/v)中,以十倍稀釋法對其進行梯度稀釋后,吸取200 μL稀釋度為10-5、10-6、10-7的稀釋液涂布于已準備好的M17和MRS固體平板培養基上。將涂布的平皿放入厭氧培養罐中(氣體條件:CO2∶H2∶N2=10∶10∶80),并置于 30 ℃培養48~72 h,菌落形成后觀察其形態。依據之前報道中乳酸菌的菌落形態特征,將疑似為乳酸菌的菌落做標記,并在無菌操作臺中用接種針或接種環挑取標記好的菌落分別相應接種于MRS和M17液體培養基中,30℃厭氧培養24 h。繼續傳代后挑取適量菌體進行革蘭氏染色試驗(涂片、固定、染色、脫色),在顯微鏡下仔細觀察并記錄細胞形態和細菌排列方式。將革蘭氏染色陽性的菌株暫定為乳酸菌[14],并進一步純化培養保存。
1.3.2 乳酸菌基因組DNA提取及16S rRNA測序
將純化好的菌株采用CTAB法提取總DNA[15]。并利用PCR擴增其16S rRNA基因片段。擴增引物采用通用引物:
正向引物為27F:5'-AGAGTTTGATCCTGGCTCAG-3’;
反向引物為1495R:5'-CTACGGCTACCTTCTTACGA-3’。
PCR反應體系見表2:

表2 PCR反應體系(25μL)Table 2 PCR reaction system(25μL)

取5 μL PCR產物和2 μL上樣緩沖液(6×Loading Buffer)混合,采用1.0%瓊脂糖凝膠進行電泳檢測,電壓5 V/cm,電泳液為0.5×TBE。電泳30 min后用凝膠成像儀觀察結果,片段長度約1 500bp的陽性產物經純化后送上海桑尼生物技術有限公司進行測序。測定的序列用BLAST在GenBank中搜索相近序列(http://www.ncbi.nlm.nih.gov/blast/)。將測序菌株與標準菌株的16S rDNA序列首先使用ClustalX[16]將序列進行完全比對,如果菌株與模式菌株的同源性大于99%,則可鑒定此菌株與模式菌株為同一菌株,然后用Neighbor-joining法[17]取得序列的進化距離。使用MEGA5軟件[18]作出系統進化樹,數據自展重抽樣次數1 000次。所得菌株16S rRNA序列全部提交GenBank數據庫。
1.4 454焦磷酸測序材料的獲取和數據分析方法
1.4.1 基因組DNA提取
采用破碎法提取2份樣品中基因組DNA。具體步驟如下:將采集的原始樣品在漩渦振蕩器上振蕩30 s混勻,取800 μL樣品于1.5 mL滅菌 EP管中。12 000 g離心10 min,棄去上清液,沉淀中加1 mL PBS緩沖液,漩渦振起。12 000 g離心10 min,棄去上清,沉淀加800 μL Tris-EDTA緩沖液,漩渦振勻后轉至裝有玻璃珠的破碎管中,在破碎儀上破碎3次。在破碎液中加入60 μL SDS,振勻后在4℃放置5 min。12 000 g離心10 min,轉移上清液到新的1.5 mL滅菌 EP管中。加入100 μL NaCl溶液(5 mol/L),80 μL CTAB(1%,pH=8.0),65 ℃水浴 20 min。加 V(酚)∶V(三氯甲烷)∶V(異戊醇)=25∶24∶1至約1.5 mL處,顛倒混勻,靜置1 min,重復3次。4℃,12 000 g離心10 min。吸取上清液轉移至新的1.5 mL滅菌EP管中,加入等體積的三氯甲烷異戊醇,室溫12 000 g離心5 min。收集上清液,加入0.1倍體積的醋酸鈉(3 mol/L),顛倒混勻,加入1倍體積的冰異戊醇,輕柔混勻后靜置20 min。12 000 g離心5 min,棄去上清液,加500 μL 70%的乙醇洗滌沉淀。棄去上清液,沉淀自然風干。沉淀加50 μL Tris-EDTA緩沖溶液回溶,冷藏備用。
1.4.2 基因組DNA擴增及焦磷酸測序
DNA擴增采用的是細菌16s rDNA V1-V3區通用引物。具體的PCR擴增引物:
正向引物為27F:5'-AGAGTTTGATCCTGGCTCAG-3’;
反向引物為533R:5'-TTACCGCGGCTGCTGGCAC-3’。
反應體系見表3:
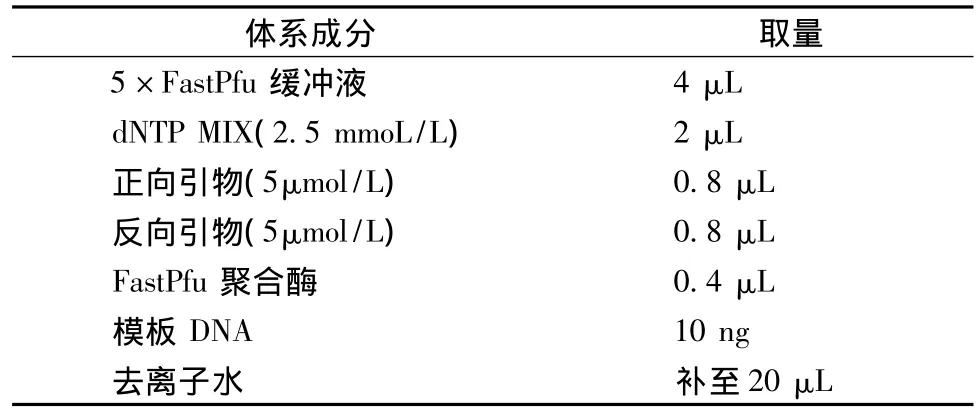
表3 PCR反應體系(20 μL)Table 3 PCR reaction system(20μL)
PCR反應產物用2%瓊脂糖凝膠電泳檢測合格后用于測序,焦磷酸測序按照454 Roche GS-FLX的標準流程進行。454焦磷酸測序結果已上傳到MG-RAST網站,Ru12和Ru13樣品編號分別為4552295.3和4552296.3。
1.4.3 數據分析
根據以下標準對原始測序序列進行篩選:
(1)長度篩選(篩選出原始序列中長度大于300 bp的序列)。
(2)引物匹配(blastn 的方法,W=4,e=0.1,找到序列上與引物匹配的片段,確定上下游引物的位置)。
(3)可變區篩選(找到引物位置后,認為兩端引物之間的片段為可變區,選出其中長度大于300 bp的片段)。
(4)tag匹配(tag按照樣本-tag編號篩選,不允許錯配,且必須為首7位)。
(5)質量控制(整條序列上質量大于20的堿基所占比例須高于93%)。
經過以上5步篩選,認為提取出的序列是長度、質量、引物、tag均有效的高質量序列。通過序列的聚類以給定相似度0.97水平上劃分操作分類單元(operational taxonomic units,OTUs),選取每一 OTU 的代表性序列進行注釋。使用本地BLAST的方法比對,采用Ribosomal Database數據庫。
2 結果與分析
2.1 分離菌株的鑒定
采集自俄羅斯卡爾梅克共和國埃利斯塔市郊的2份發酵蔬菜樣品共分離鑒定出19株乳酸菌,分屬于植物乳桿菌屬(14株)、片球菌屬(3株)和明串珠菌屬(2株),其中Ru12樣品分離鑒定出11株,Ru13樣品為8株。對這些分離株進行16S rDNA序列分析并構建系統發育樹,參見圖1。通過BLAST序列比對分析和系統進化樹分析可知:所有19株菌初步歸于三大群,其中有7株菌與Lactobacillus plantarum(T)AJ965482同源性在99%以上,將其歸屬為Lactobacillus plantarum;有1株菌與 Lactobacillus brevis ATCC 367同源性為100%,將其歸屬為Lactobacillus brevis;有2株菌與Lactobacillus delbrueckii(T)ATCC 11842同源性在99%以上,將這2株菌歸屬為Lactobacillus delbrueckii;有1株菌與 Lactobacillus helveticus(T)AM113779同源性在99%以上,將其歸為Lactobacillus helveticus;有 1株菌與 Lactobacillus kefiranofaciens AM113782同源性在99%以上,將其歸屬為Lactobacillus kefiranofaciens;有 2株菌與 Lactobacillus kefiri(T)AJ621553同源性在99%以上,將這2株菌歸屬為Lactobacillus kefiri;有1株菌與Leuconostoc lactis(T)AB023968同源性在99%以上,將其歸屬為Leuconostoc lactis;有1株菌與 Leuconostoc mesenteroides(T)ATCC 8293同源性為100%,將其歸屬為Leuconostoc mesenteroides;有2株菌與Pediococcus ethanolidurans AY956789同源性大于99%,將這2株菌歸屬為Pediococcus ethanolidurans;有1株菌與Pediococcus pentosaceus ATCC 25745同源性大于99%,將其歸屬為Pediococcus pentosaceus。對于上述分離菌株的生物學特性還需要后續的實驗進行分析和驗證,從而進一步篩選適合發酵蔬菜工業化生產的菌株。

圖1 19株待測菌株與參考菌株16S rDNA序列系統發育樹Fig.1 Phylogenetic tree of the 16S rDNA of reference strains and nineteen isolates
2.2 樣品中細菌豐度和多樣性分析
通過454焦磷酸測序,Ru12發酵蔬菜共得到10 869條高質量序列,Ru13樣品為12 204條,經過PyNAST alignment和100%序列鑒定聚類(sequence identity clustering)分析后,共得到2 664條代表性序列。繼而在97%相似度水平上劃分OTU,Ru12和Ru13樣品分別發現148個和123個細菌OTU。
由圖2可知,2份發酵蔬菜的稀疏曲線均未進入平臺期,這表明隨著測序量的增加新的種系型有可能被發現,但同時Shannon曲線已經飽和,表明隨著測序量的增加細菌的多樣性已經不再隨之發生變化。
對于一個試驗中至少需要多少條高質量的16S rRNA序列才能滿足后續的科研分析一直是科研人員比較關心的問題。既往研究表明,如果只是在門的水平上分析樣品中微生物組成,那么幾百條高質量的454焦磷酸測序序列就足夠了,而如果要檢測樣品中多數優勢微生物的菌群結構,幾千條序列足以達到實驗目的[19]。本實驗中2個樣品測序量都已到達一萬以上,結合以往的經驗,在該測序水平上測序結果已能夠充分揭示2份樣品中細菌的組成。
通過chao1指數對樣品的豐度進行比較,Ru12和Ru13兩份樣品的chao1指數分別為233.8和171.0,表明Ru12樣品中細菌的豐度比Ru13樣品要高。通過simpson指數對樣品的多樣性進行比較分析,Ru12和Ru13兩份樣品的simpson指數分別為0.527和0.534,這表明2份樣品中細菌的多樣性差別不大。
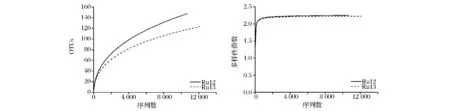
圖2 兩份發酵蔬菜樣品的稀疏曲線和Shannon曲線Fig.2 Rarefaction analysis and shannon diversity index of two fermented vegetable simples
2.3 樣品中細菌相對含量的比較分析
通過在97%相似度上劃分OTU,每一OTU選取出現次數最多的序列為代表性序列,采用本地BLAST的方法進行注釋,在門的水平上,兩份樣品都主要由硬壁菌門(Firmicutes)、變形菌門(Proteobacteria)、放線菌門(Actinobacteria)和擬桿菌門(Bacteroidetes)組成,在Ru12樣品中,上述門類細菌的含量分別為96.26%、3.66%、0.04%、0.02%,在Ru13樣品中,其含量分別為 97.64%、2.27%、0.07%、0.02%,參見圖3、圖4。
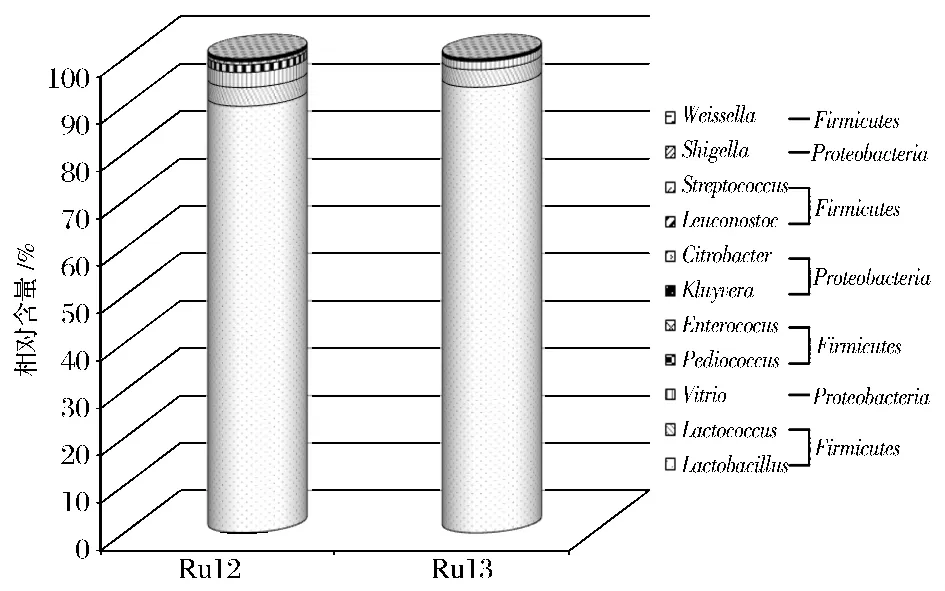
圖3 兩份發酵蔬菜樣品中細菌的組成Fig.3 Compositions of bacteria in two fermented vegetable samples
由圖3、圖4可知,在屬的水平上,2份樣品中含量最高的細菌屬主要包括乳桿菌屬(Lactobacillus)、乳球菌屬(Lactococcus)、弧菌屬(Vibrio)、片球菌屬(Pediococcus)、腸球菌屬(Enterococcus)、克魯維菌屬(Kluyvera)等。在Ru12樣品中這些菌屬的相對含量依次為 89.41%、3.95%、3.14%、1.61%、0.64%、0.08%,在Ru13樣品中的相對含量依次為93.65%、3.69%、1.69%、0.7%、0.08%、0.21%。由此可見,在2份樣品中,占據支配地位的菌屬都是乳桿菌屬(Lactobacillus),其次為乳球菌屬(Lactococcus)和弧菌屬(Vibrio)。上述實驗結果也與乳酸菌分離鑒定中分離到的乳桿菌屬的數量占到絕大多數的結果相吻合。通過上述結果我們還發現了一個有意思的現象,在2份樣品中弧菌屬(Vibrio)都占據了其中相當大的比例,而該菌屬的很多種類是具有致病性的,對于樣品中弧菌屬的來源我們還需進一步研究。
Ji[20]等采用454焦磷酸測序技術對韓國傳統發酵蔬菜Kimchi中的微生物組成進行了分析,結果發現在Kimchi中最主要的細菌屬依次為明串珠菌屬(Leuconostoc)、乳桿菌屬(Lactobacillus)和魏斯氏菌屬(Weissella),而明串珠菌屬(Leuconostoc)和魏斯氏菌屬(Weissella)在本文所研究的樣品中相對含量都很低。這說明不同地區、不同發酵蔬菜種類中微生物的構成存在很大差異,可能正是這些微生物構成的差異造就了不同發酵蔬菜所特有的口感和風味。
3 結論
本研究通過對采集自俄羅斯卡爾梅克共和國埃利斯塔市郊的2份發酵蔬菜采用純培養結合16S rRNA序列比對分析的方法,共從2份樣品中分離出19株乳酸菌,并對其進行了準確的鑒定和分型,鑒定結果為7株Lactobacillus plantarum;1株Lactobacillus brevis;2株Lactobacillus delbrueckii;1株Lactobacillus helveticus;1株Lactobacillus kefiranofaciens;2株Lactobacillus kefiri;1株Leuconostoc lactis;1株Leuconostoc mesenteroides;2株Pediococcus ethanolidurans;1株Pediococcus pentosaceus。同時運用454焦磷酸測序技術,本研究從宏基因組水平分析了2份發酵蔬菜樣品中細菌的豐度和多樣性,發現隸屬于硬壁菌門(Firmicutes)的乳桿菌屬(Lactobacillus)和乳球菌屬(Lactococcus)是2份樣品中的優勢菌屬,這也證實了之前通過純培養方法得到的部分研究結論。此外,通過454焦磷酸測序技術我們對發酵蔬菜中細菌的豐度和多樣性有了一個更加全面深入的認識。例如,我們在研究中還發現了樣品中部分有害菌屬的存在,這是之前研究未曾報道的現象。上述研究成果對于發酵蔬菜工業化生產的發展具有積極的促進作用。
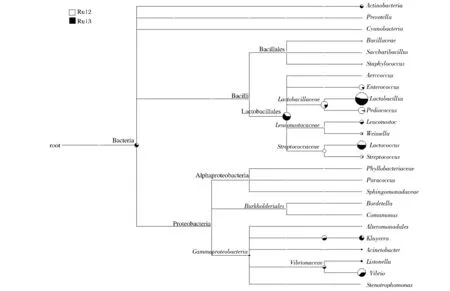
圖4 兩份發酵蔬菜樣品中細菌相對含量的比較Fig.4 Relative abundance of bacteria in two fermented vegetable samples
[1] Park J M,Shin J H,Lee D W,et al.Identification of the lactic acid bacteria in kimchi according to initial and overripened fermentation using PCR and 16S rRNA gene sequence analysis[J].Food Science and Biotechnology,2010,19(2):541-546.
[2] Shin M S,Han S K,Ryu J S,et al.Isolation and partial characterization of a bacteriocin produced by Pediococcus pentosaceus K23-2 isolated from Kimchi[J].Journal of Applied Microbiology,2008,105(2):331-339.
[3] Kim M,Chun J.Bacterial community structure in kimchi,a Korean fermented vegetable food,as revealed by 16S rRNA gene analysis[J].International Journal of Food Microbiology,2005,103(1):91-96.
[4] Ha J H.Changes of free sugars in Kimchi during fermentation Jae-Ho Ha,Wooderck S.Hawer,Young-Jin Kim and Young-Jung Nam[J].Changes,1989,21(5):633-638.
[5] Amann R I,Ludwig W,Schleifer K H.Phylogenetic identification and in situ detection of individual microbial cells without cultivation[J].Microbiological Reviews,1995,59(1):143-169.
[6] Quigley L,O'Sullivan O,Beresford T P,et al.Highthroughput sequencing for detection of subpopulations of bacteria not previously associated with artisanal cheeses[J].Applied and Environmental Microbiology,2012,78(16):5 717-5 723.
[7] Masoud W,Takamiya M,Vogensen F K,et al.Characterization of bacterial populations in Danish raw milk cheeses made with different starter cultures by denaturating gradient gel electrophoresis and pyrosequencing[J].International Dairy Journal,2011,21(3):142-148.
[8] Dobson A,O'Sullivan O,Cotter P D,et al.High-throughput sequence-based analysis of the bacterial composition of kefir and an associated kefir grain[J].FEMS Microbiology Letters,2011,320(1):56-62.
[9] Leite A M O,Mayo B,Rachid C,et al.Assessment of the microbial diversity of Brazilian kefir grains by PCR-DGGE and pyrosequencing analysis[J].Food Microbiology,2012,31(2):215-221.
[10] Jung M J,Nam Y D,Roh S W,et al.Unexpected convergence of fungal and bacterial communities during fermentation of traditional Korean alcoholic beverages inoculated with various natural starters[J].Food Microbiology,2012,30(1):112-123.
[11] 小崎道雄,內村泰,岡田早苗.乳酸菌実験マニュアル[J].東京:朝倉書店,1992:36-59.
[12] 辨野義己.乳酸菌の分類[J].微生物,1990,6:3-14.
[13] Sneath P H A,Mair N S,Sharpe M E,et al.Bereys manual of systematic bactetiology.Vol.2[M].Williams 8L Wilkins,1986.
[14] 凌代文.乳酸細菌分類鑒定及實驗方法[M].北京:中國輕工業出版社,1999.
[15] F奧斯伯,R布倫特(顏子穎,王海林,譯).精編分子生物學實驗指南[M].北京:科學出版社,1998.
[16] Thompson J D,Gibson T J,Plewniak F,et al.The CLUSTAL_X windows interface:flexible strategies for multiple sequence alignment aided by quality analysis tools[J].Nucleic Acids Research,1997,25(24):4 876-4 882.
[17] Saitou N,Nei M.The neighbor-joining method:a new method for reconstructing phylogenetic trees[J].Molecular Biology and Evolution,1987,4(4):406-425.
[18] Tamura K,Dudley J,Nei M,et al.MEGA4:molecular evolutionary genetics analysis(MEGA)software version 4.0[J].Molecular Biology and Evolution,2007,24(8):1 596-1 599.
[19] Hamady M,Knight R.Microbial community profiling for human microbiome projects:Tools,techniques,and challenges[J].Genome Research,2009,19(7):1 141-1 152.
[20] Jung J Y,Lee S H,Kim J M,et al.Metagenomic analysis of kimchi,a traditional Korean fermented food[J].Applied and Environmental Microbiology,2011,77(7):2 264-2 274.
