活性炭富集-原子吸收分光光度法測定金
2014-12-16 07:51:40羅永紅韋真周
濕法冶金 2014年4期
羅永紅,韋真周
(廣西冶金研究院,廣西 南寧 530023)
用活性炭富集-原子吸收分光光度法測定金方法簡便,勞動強度小,分析結果準確性好,能滿足生產和科研需要[1-3]。但該法的準確性受多種因素影響[4-5],為進一步提高靈敏度及準確性,結合工作實際,探討了該法中影響測定結果準確性的幾個因素。
1 材料與方法
1.1 儀器與試劑
試驗儀器:AL204型電子天平,梅特勒-托利多儀器(上海)有限公司;KSW-5-16S型馬弗爐,上海滬越實驗儀器有限公司;ACS-6A型電子計重稱,上海友聲衡器有限公司;TAS-986F型原子吸收分光光度計,北京通用儀器有限責任公司;金空心陰極燈,儀器工作參數見表1。

表1 原子吸收分光光度計工作參數
試劑:硝酸,鹽酸,氟化鈉,氟化氫銨,氯化鈉,高錳酸鉀,均為分析純;硫脲,10g/L,分析純。
金標準溶液儲備液:用電子天平準確稱取0.100 0g純金(99.99%)置于100mL燒杯中,加入10mL王水,置于電熱板上水浴加熱至金完全溶解,加入15滴250g/L氯化鈉溶液,繼續水浴加熱蒸干至無酸味,加2mL濃鹽酸重復蒸干2次。取下冷卻,以去離子水溶解并轉入100mL容量瓶中,加2mL濃鹽酸,用水稀釋至刻度,搖勻,于陰涼處密封保存。溶液中金質量濃度為1mg/mL。
紙漿:由定性濾紙在水中搗碎制備。
活性炭:將200目活性炭在50g/L氟化氫銨的王水(1+4)溶液中浸泡1周,抽濾后,以0.24 mol/L鹽酸和去離子水洗凈。
吸附柱:內徑32mm,高60mm。
1.2 校準曲線
取金標準儲備液10mL,用8mol/L鹽酸定容于100mL容量瓶中,此溶液為金標準溶液,金質量濃度為100μg/mL。
分別取0,0.5,1.0,2.0,3.0,4.0mL 金標準溶液于100mL容量瓶中,加入2mL鹽酸溶液(6 mol/L),10mL硫脲溶液(10g/L),用水稀釋至刻度。此標準系列溶液金質量濃度分別為0,0.5,1.0,2.0,3.0,4.0μg/mL。
2 試驗方法
視含金量,用電子天平稱取10~30g試樣置于灰皿中,放入馬弗爐中在700℃下焙燒1h,冷卻后取出,轉入400mL燒杯中,用水沖洗灰皿2~3次。移至通風櫥中,加入100mL王水(1+1),蓋上表皿,于電熱板上加熱并保持微沸30~60min。停止加熱后用溫熱水沖洗表皿及杯壁,稀釋至200~250mL,冷卻至40~50℃時過濾。
于裝有活動板的吸附柱上加入直徑為30 mm的濾紙,倒入紙漿,抽干,使紙漿高度為2~3 mm,再倒入活性炭紙漿,抽干,厚度視待測樣品的含金量而定。活性炭紙漿上再倒一層紙漿,壓平,抽干,以完全覆蓋住活性炭紙漿為宜。用水沖洗柱壁,裝上布氏漏斗,布氏漏斗中鋪一層7cm濾紙,用水潤濕,倒入少許紙漿,抽干。試樣溶液連同殘渣一起倒入布氏漏斗,抽濾。抽濾后,以0.24mol/L溫熱鹽酸沖洗燒杯壁2~3次,沖洗漏斗上殘渣7~8次。去掉布氏漏斗,再次沖洗4~5次吸附柱,然后以40~50℃去離子水沖洗吸附柱10~12次。停止抽濾后取下活性炭紙漿塊,置于50mL坩堝中,在電熱板上碳化至無煙為止,再放入馬弗爐中于700℃下灰化至完全。取出冷卻后,在坩堝中加入2mL王水,電熱板上水浴蒸至濕鹽狀(約0.5mL,不宜過干),加入硫脲溶液(按每10mL定容體積加1mL),水洗坩堝,移入容量瓶中,按儀器條件進行測定。
礦石中金的質量分數計算公式為

式中:w(Au)為金的質量分數,g/t;ρ1為工作曲線上查得的試液金質量濃度,μg/mL;ρ0為工作曲線上查得的空白試液金質量濃度,μg/mL;V為試液定容體積,mL;V1為試液分取體積,mL;V2為試液稀釋體積,mL;m為試樣質量,g。
3 試驗結果與討論
3.1 焙燒方式的影響
對同一樣品稱取各兩份(銅精礦,硫鐵礦):一份低溫下放入馬弗爐中,爐門不完全關閉,升溫至設定溫度(700℃)后保持1h,關閉電源,待其冷卻后取出;另一份先升溫至400℃左右,攪拌至顏色均勻后再升溫至700℃保持1h。焙燒以松散不板結、易溶于王水為宜。兩種焙燒樣品分別按試驗方法進行測定,試驗結果如圖1所示。可以看出:焙燒過程中加以攪拌,焙燒樣品的測定值與推薦值相近;而未攪拌的樣品,其測定值明顯偏低。同時,未攪拌焙燒樣品均存在板結、不易溶解現象。硫鐵礦中金的測定結果偏低可能是灰皿底部的C、S未完全燒盡的緣故。
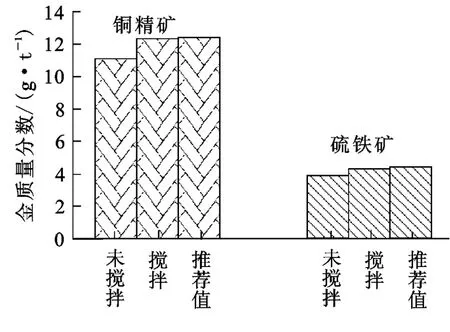
圖1 焙燒方式對測定結果的影響
3.2 金的溶解的影響
顯微鏡觀察結果表明,試樣中的金通常以3種方式存在:二氧化硅鑲嵌共生物,金屬顆粒,片狀[4-5]。金被硅酸鹽或二氧化硅包裹時溶解不完全,需加氯化鈉、氟化鈉、高錳酸鉀助溶,加助溶劑后再以王水(1+1)溶解。
取等量試樣(磨碎至200目左右),一份加入助溶劑氯化鈉、氟化鈉、高錳酸鉀,另一份不加助溶劑,都加100mL王水(1+1)溶解后,按試驗方法進行測定。試驗結果如圖2所示。

圖2 助溶劑對結果測定結果的影響
由圖2看出:不加助溶劑時,測定結果偏低;而加入助溶劑后,測定重復性和平行性均較好,與推薦值相符合。
通常情況下,金在王水中的反應如下[6]:
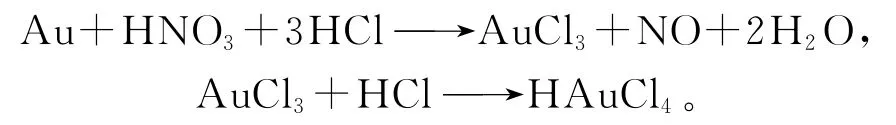
當氯離子大量存在時,硝酸的氧化性更強。酸性條件下,高錳酸鉀與氯化鈉發生如下反應:

反應中生成的氯氣能更好地溶解試樣中的金,因此,加入氟化鈉、氯化鈉、高錳酸鉀,不僅促進金的溶解,也減少了王水用量,可避免濃王水溶樣引起的劇烈反應。
3.3 活性炭紙漿厚度的影響

對于動態吸附而言,活性炭紙漿吸附層厚度,試液通過活性炭紙漿吸附層速率,活性炭紙漿層密度都會影響金的吸附[7-9]。后兩者相同時,活性炭紙漿吸附層厚度對金吸附試驗結果見表2,吸附原液為純的AuCl-4溶液。

表2 活性炭紙漿厚度與金吸附量的關系
實際試樣中往往含有其他離子,如鐵、銅、錫等,這些離子也會吸附在活性炭上,影響金的吸附。根據實際工作經驗,建議活性炭與紙漿的質量比為1∶2的活性炭紙漿厚度以7mm左右為宜。
3.4 活性炭灰化程度的影響
取等量試樣2份,按試驗方法進行測定。試樣碳化后,放入馬弗爐中在700℃下進行灰化,灰化程度對金測定的影響試驗結果如圖3所示。

圖3 活性炭灰化程度對金的測定結果
由圖3看出,活性炭未完全灰化,測定結果偏低,這可能是未完全灰化的活性炭中的金不能被王水完全溶解所致。
4 結論
試驗比較了試樣焙燒、溶解、活性炭紙漿厚度、灰化程度對活性炭富集-原子吸收分光光度法測定金的影響,結果表明:
1)銅精礦、硫鐵礦試樣焙燒時,需在400℃下攪拌至顏色均勻,再繼續在700℃下焙燒,避免因焙燒不完全造成測定結果偏低。
2)以氟化鈉、高錳酸鉀、氯化鈉作助溶劑,能更好地溶解硅酸鹽或二氧化硅包裹的金。
3)活性炭紙漿厚度以7mm為宜;測定時活性炭需完全灰化。
[1]梁慧貞,祁之軍.活性炭吸附富集測定礦石中的金[J].科技風,2010(16):224.
[2]董喜明.活性炭富集碘量法測定金的探討[J].黃金,2000,21(7):43-46.
[3]韓朝輝.原子吸收光度法測定金的幾個問題討論[J].甘肅冶金,2010,32(6):107-109.
[4]朱盈權,李俊義.金的分析現狀[J].華中師院學報:自然科學版,1979,(1):59-72.
[5]王民權.關于活性炭-碘量法試金中幾個問題的初步探討[J].黃金,1983,7(3):58-61.
[6]郭愛武,曹曉武,杜作朋,等.氯化銨焙燒金銻共生礦中金的測定[J].光譜實驗室,2010,27(4):1557-1559.
[7]康澤彥.活性炭富集原子吸收分光光度法測定金[J].新疆地質,2005,23(3):316-317.
[8]李志明,劉桂芬.原子吸收光譜法測定金銀樣品前處理的討論[J].分析測試技術與儀器,2005,11(1):71-74.
[9]張秀麗.影響活性炭吸附原子吸收光譜法測定金的因素[J].新疆有色金屬,2009(5):36-37.
