硅基高交聯磺化PS-DVB離子交換劑的研究
2015-01-01 02:34:56蔡大川鄭家概潘燦盛曾志堅左雄軍
分析測試學報 2015年11期
張 飛,蔡大川,鄭家概,潘燦盛,曾志堅,左雄軍*
(1.中國廣州分析測試中心 廣東省化學危害應急檢測技術重點實驗室,廣東 廣州 510070;2.華南師范大學 實驗中心,廣東 廣州 510006;3.華南師范大學 化學與環境學院,廣東 廣州 510006)
離子色譜分離通常采用酸、堿或鹽溶液作為流動相,因此對離子交換劑的耐酸、堿和鹽穩定性要求很高,一般需采用高穩定性和合適擔載量的聚合物作為離子交換劑[1]。其中以苯乙烯-二乙烯基苯(PS-DVB)聚合物為基質的離子交換劑是聚合物基質中最常用的離子交換劑[2-4]。該類離子交換劑可在pH 1~14范圍內使用,能解決眾多離子分離的問題。但其擔載量太高時,聚合物質脆,裝柱時易碎,從而造成堵塞;而擔載量太小則溶脹性和收縮性較大,不易獲得穩定的柱床層,還易被微生物消化,不易保存[5-6]。以硅膠為基質的鍵合相離子交換劑可以克服聚合物基質離子交換劑的上述缺點,但其耐酸、堿和鹽的穩定性不足,只能在pH 2~8范圍內使用[7-8],故很多離子樣品難以用硅膠鍵合相離子交換劑分離。包埋共聚法(Encapsulated copolymerization)[9-10]、逐層疊加方法(Layer by layer,LBL)[11]以及基于雙官能團的虎-克交聯法[12]等均可以制備不同聚合物擔載量的硅基聚合物離子交換劑。這類離子交換劑的耐酸、堿、鹽穩定性與聚合物類型、聚合物在硅小球表面的覆蓋程度有關。聚合物越穩定,離子交換劑的耐酸、堿和鹽穩定性越好;聚合物擔載量越大,離子交換劑越穩定。但隨著離子交換劑表面聚合物擔載量的增加,樣品在離子交換劑內的傳質阻力增大,柱壓顯著上升。升高分離溫度不僅可以加快溶質在離子交換劑內的傳質速率,還可以降低柱壓和改善柱效[13-16]。本文結合上述兩種離子交換劑的優點,采用包埋共聚法制備了剛性、高穩定性的硅基高交聯磺化PS-DVB交換劑。該離子交換劑將聚苯乙烯層包裹在硅基鍵合相表面,可較好地結合硅基材質和高分子材質的優點,克服二者各自的不足。覆蓋在硅基鍵合相表面的聚苯乙烯層可以阻止酸或堿對內層硅膠的侵蝕,而硅膠可為外層的高分子離子交換劑提供很好的支撐。
1 實驗部分
1.1 儀器與試劑
CGY-100 B裝柱機(北京福思源機械加工部);LC-10A液相色譜(蘇州島津公司):兩臺LC-10AD高壓泵、Rebdyne 7 725六通進樣閥(20 μL)、紫外-可見光檢測器和HW-2 000色譜工作站、熱重差示掃描量熱儀(美國Perkin-Elmer公司)。
硅小球(粒徑5 μm,實驗室自制);鹽酸、過氧化苯甲酰、醋酸銨(廣州化學試劑廠),四甲基二乙烯基二硅氧烷(廣州聚成兆業有機硅公司),甲苯、甲醇、無水乙醇、氫氧化鈉、無水硫酸鈉、氯仿(天津市大茂化學試劑廠),苯乙烯(天津市福晨化學試劑廠),二乙烯基苯(Aldrich公司),乙醚(衡陽市凱信化工試劑有限公司),氯磺酸(廣州金華大化學試劑有限公司),三乙胺(上海萬翔化工原料貿易有限公司),醋酸(天津市富宇精細化工有限公司),以上試劑均為分析純;乙腈(色譜純,Fisher公司),實驗用水為去離子水。
1.2 硅基高交聯磺化PS-DVB離子交換劑的制備
苯乙烯-二乙烯基苯混合單體溶液:分別量取100 mL苯乙烯和50 mL二乙烯基苯,混勻,配成混合單體,再用4 mol/L NaOH處理此混合單體溶液3次,每次150 mL,以除去混合單體溶液中的阻聚劑對苯二酚。再向混合溶液中加4 g無水硫酸鈉,靜置除水1 d,過濾除去混合溶液中的硫酸鈉,-20℃冷凍保存,備用。
硅基高交聯磺化PS-DVB離子交換劑的制備過程如下:
①硅小球的活化:稱量60 g硅小球于500 mL三頸燒瓶中,加入360 mL 20%HCl,攪拌回流24 h,冷卻后過濾活化硅小球,用蒸餾水洗至中性,無水乙醇洗滌2次,每次100 mL,105℃真空干燥24 h,待用。
②硅小球的鍵合:將活化的硅小球置于500 mL三頸燒瓶中,加入234 mL無水甲苯和42 mL四甲基二乙烯基二硅氧烷,攪拌回流24 h。冷卻至室溫后,依次用無水乙醇、水、無水乙醇洗滌2次,每次100 mL,105℃真空干燥24 h,制得乙烯基硅烷鍵合相,待用。
③包埋共聚反應:在250 mL圓底燒瓶中,加入15 g乙烯基硅烷鍵合相,再加入一定量的苯乙烯-二乙烯基苯混合單體溶液(用乙醚補足100 mL)和0.5 g過氧化苯甲酰引發劑。超聲振蕩15 min后,45℃下旋轉蒸發除去乙醚,再將反應物置于90℃下恒溫4 h。依次用甲苯、無水乙醇洗滌產物3次,每次100 mL,除去未發生聚合的苯乙烯-二乙烯基苯單體以及未發生包埋的聚合物,105℃真空干燥12 h,制得硅基高交聯PS-DVB。
④磺化反應:在125 mL三頸瓶中,加入5 g硅基高交聯PS-DVB和50 mL無水氯仿,置于冰水浴中。攪拌反應液,滴加5 mL氯磺酸后,繼續反應4 h。再加入6 mol/L NaOH溶液,中和未反應的氯磺酸,并調至pH 14。靜置4 h后減壓抽濾,用水洗滌產物直至濾液呈中性,再用無水乙醇洗滌2次,每次100 mL,產物置于105℃真空干燥12 h。
1.3 色譜柱裝填
稱取“1.2”制備的離子交換劑4.2 g于100 mL三角瓶中,向其中加入25 mL 1,4-二氧六環和25 mL氯仿,超聲10 min制得離子交換劑勻漿液。再用甲醇作頂替液,于60~80 MPa壓力下填充到250 mm×4.6 mm不銹鋼管柱內。將裝填好的色譜柱連接到色譜儀器中,流速1.0 mL/min,用甲醇和水分別沖洗2 h。
1.4 色譜條件
色譜柱(250 mm×4.6 mm I.D.)。流動相為30 mmol/L NH4Ac-乙腈-三乙胺(95∶5∶0.1),使用時根據實際情況用醋酸調至所需pH值;流速:0.8 mL/min;UV:254 nm;溫度:25℃。
2 結果與討論
2.1 離子交換劑的制備與表征
2.1.1 離子交換劑的制備 離子交換劑的制備過程如“1.2”所述。實驗以活化硅小球為基質,通過硅烷化試劑四甲基二乙烯基二硅氧烷,在硅小球表面通過化學鍵合引入乙烯基;以苯乙烯為聚合單體,二乙烯基苯為交聯劑,通過引發劑過氧化苯甲酰引發聚合,在硅基鍵合相表面包裹一層PS-DVB薄膜;再將得到的填料與氯磺酸發生磺化反應,引入磺酸基制得強陽離子交換劑,制備路線見圖1。
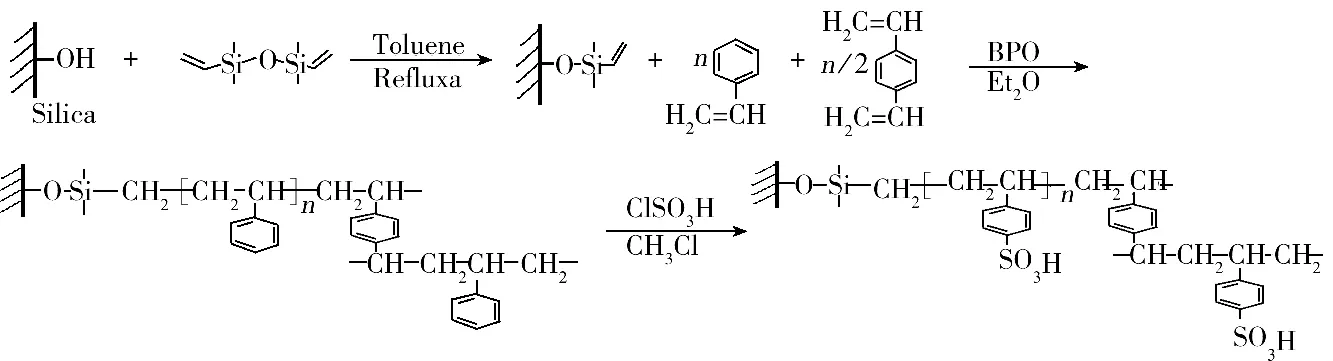
圖1 硅基高交聯磺化PS-DVB離子交換劑的制備路線Fig.1 Preparation of a silica-based sulfonated hypercrosslinked polystyrene-divinylbenzene strong cation-exchanger
2.1.2 4種硅基高交聯磺化PS-DVB離子交換劑擔載量的計算 聚合物擔載量是指硅基高交聯磺化PS-DVB離子交換劑中聚苯乙烯的含量。在包埋共聚反應過程中,通過調節乙烯基硅烷鍵合相與苯乙烯-二乙烯基苯混合單體的反應比例(15 g∶10 mL;15 g∶8.5 mL;15 g∶7.5 mL;15 g∶5.5 mL),制得4種聚合物擔載量的硅基高交聯PS-DVB。采用熱重分析法計算離子交換劑的擔載量依次為33%,24%,19%和12%。
2.1.3 紅外光譜表征 采用紅外光譜法分別對活化硅小球、乙烯基硅烷鍵合相、硅基PS-DVB共聚物和硅基高交聯磺化PS-DVB離子交換劑(擔載量為19%)進行表征,結果見圖2。從漫反射紅外光譜可見,活化硅小球(圖2A-a)加入甲苯和四甲基二乙烯基二硅氧烷后制得乙烯基硅烷鍵合相(圖2A-b)后,在1 640 cm-1處出現—CH‖CH2伸縮振動,3 068 cm-1處出現C‖C—H的伸縮振動,表明硅小球表面鍵合上了乙烯基。再經包埋共聚反應后在1 605,1 450 cm-1處產生苯環C‖C雙鍵伸縮振動(圖2A-c),證明硅小球表面已吸附上聚苯乙烯聚合物,形成了硅基PS-DVB共聚物。對硅基PSDVB共聚物進行磺化反應后,用KBr壓片透射紅外光譜可觀察到在1 190,1 068 cm-1處出現磺酸基特征吸收峰(圖2B-d),證明硅基PS-DVB共聚物上引入了磺酸基。離子交換劑經6 mol/L NaOH溶液處理后,苯環的特征伸縮振動峰(1 605 cm-1)依然清晰可見,表明該離子交換劑具有較好的耐酸堿穩定性。
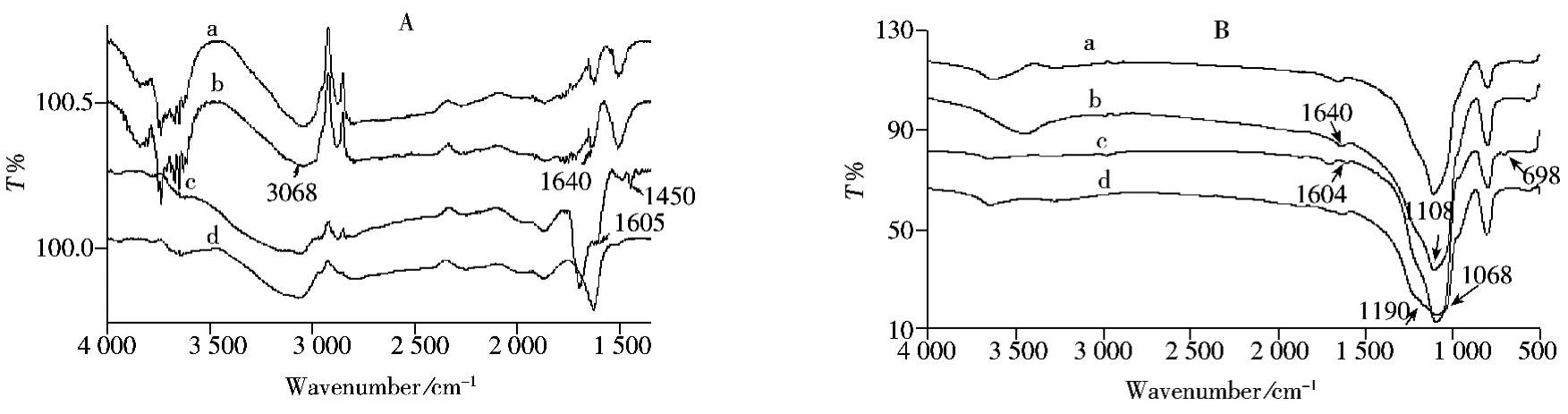
圖2 離子交換劑的漫反射紅外光譜(A)與KBr壓片透射紅外光譜(B)Fig.2 Infrared diffuse reflectance spectra(A)and infrared transmission method of KBr pellets(B)
2.2 色譜性能的考察
通常情況下,聚合物擔載量越大,溶質在離子交換劑內的傳質效率越低。如果聚合物擔載量太小,柱穩定性會迅速降低。因此,優化聚合物擔載量是制備性能較好的高交聯硅基強陽離子交換劑的關鍵之一。本研究合成了4種聚合物擔載量的硅基高交聯強陽離子交換劑,探討聚合物擔載量對離子交換劑性能的影響。
2.2.1 聚合物擔載量對柱壓的影響 色譜柱的柱滲透性與聚合物的擔載量密切相關。在流動相為30 mmol/L NH4Ac-乙腈-三乙胺(95∶5∶0.1,醋酸調至pH 3.6),流速0.2 mL/min,溫度25℃條件下,4種擔載量(12%,19%,24%和33%)離子交換劑的柱壓依次為4.4,6.1,10.0,25.0 MPa。表明隨著擔載量的增加,柱壓急劇升高,柱滲透性迅速下降。常溫下,由于24%和33%擔載量的離子交換劑的柱壓過高,難以實現色譜分離分析。因此,后續實驗僅對擔載量為12%和19%的離子交換劑進行研究。
2.2.2 聚合物擔載量對分離的影響 實驗考察了不同擔載量的硅基高交聯磺化PS-DVB離子交換劑對胺類和含氮雜環堿性化合物的分離情況。其中胺類化合物選擇苯胺(Aniline)和芐胺(Benzylamine)進行考察,以30 mmol/L NH4Ac-乙腈-三乙胺(95∶5∶0.1,pH 4.6)為流動相,分別用12%和19%擔載量的離子交換劑進行分離。結果表明,12%擔載量因聚合物擔載量小,未能對硅基鍵合相進行均勻覆蓋,苯胺和芐胺不能實現有效分離。而19%擔載量的離子交換劑柱效更高,苯胺和芐胺可以得到有效分離,分離度為1.2,色譜峰峰形尖銳,對稱性好。
以30 mmol/L NH4Ac-乙腈-三乙胺(95∶5∶0.1,pH 3.6)為流動相,繼續考察了12%和19%擔載量的硅基高交聯磺化PS-DVB離子交換劑對2-吡嗪羧酸(Pyrazinoic acid)、煙酸(Niacin)、苯并三氮唑(Benzotriazole)、吡嗪(Pyrazine)、吲哚(Indole)和吡啶(Pyridine)6種含氮雜環堿性化合物的分離情況,結果見圖3。12%擔載量的分離效果不理想(見圖3A),其中吡嗪、吲哚和吡啶峰拖尾嚴重,無法實現有效分離,原因可能是因擔載量小,這3種化合物除了與離子交換劑發生離子交換外,尚能和硅羥基發生較強的作用,導致峰拖尾。19%擔載量能夠很好地實現6種化合物的分離(圖3B),分離度均大于1.2。由此可見,擔載量對離子交換劑的分離性能有較大影響,擔載量過小時,溶脹性和收縮性較大,不能有效分離化合物;而擔載量過大,又會導致柱壓升高,柱滲透性降低,不利于常溫分離。因此,本文后續均采用19%擔載量的交換劑進行研究。
圖3 擔載量為12%(A)和19%(B)的離子交換劑對雜環含氮化合物的分離Fig.3 Chromatograms for separation of mixture on strong cation-exchanger of 12%(A)and 19%(B)loading
2.2.3 穩定性考察 實驗將19%擔載量的離子交換劑在流動相為30 mmol/L NH4Ac-乙腈-三乙胺(95∶5∶0.1,pH 4.6)條件下連續沖洗400 h后,再次分離分析苯胺和芐胺,結果發現,苯胺和芐胺的理論塔板數和分離度均未發生顯著變化,說明所制備的離子交換劑具有很好的穩定性。
2.3 實際應用
實驗選取3組尚未見報道分離的芳香胺類化合物,采用本文所制備的PS-DVB離子交換劑進行分離測試,3組芳香胺類化合物分別為:N,N-二甲基苯胺(N,N-Dimethylaniline)和N,N-二甲基芐胺(N,N-Dimthylbenzylamine);α-萘胺(Naphthylamine)和二苯胺(Diphenylamine);鄰硝基苯胺(o-Nitroaniline)和鄰溴苯胺(Bromoaniline)。結果表明,N,N-二甲基芐胺和N,N-二甲基苯胺的分離度為5.5,兩組分在12 min內出峰,峰形尖銳對稱(圖4A)。α-萘胺和二苯胺也能實現良好分離(圖4B),其中α-萘胺的理論塔板數為34 350,二苯胺的理論塔板數為32 805,二者的分離度為5.1,溶劑峰(Solvent peak)不干擾測定,柱效高。此外,對于不同官能團的芳胺類堿性化合物(如鄰硝基苯胺和鄰溴苯胺)也可實現較好的分離(圖4C),基線平穩,鄰硝基苯胺和鄰溴苯胺的分離度大于1.0,可用于定量分析。

圖4 19%擔載量的離子交換劑的色譜分離圖Fig.4 Chromatograms for separation of mixture on 19%loading strong cation-exchanger
3 結論
本工作以硅小球為基質,采用包埋共聚法制備了一種硅基高交聯磺化PS-DVB離子交換劑,通過分離含氮堿性化合物來考察其色譜性能。結果發現擔載量為19%的離子交換劑具有很好的穩定性,分離效果良好,有望應用于芳香胺、含氮雜環和嘧啶等堿性有機化合物的分離。
[1]Chambers K T,Fritz J S.J.Chromatogr.A,1998,797:139 -147.
[2]Liu S,Wang M M,Ai L F,Xing T,Hao Y L,Wang X S.J.Instrum.Anal.(劉珊,王曼曼,艾連峰,邢濤,郝玉蘭,王學生.分析測試學報),2013,32(5):547-552.
[3]Zhou A M,Wang Z J,Chen J L,Shen H Y,Hu M Q,Dong X Y,Xia Q H.J.Instrum.Anal.(周阿蒙,王哲君,陳君良,沈昊宇,胡美琴,董新艷,夏清華.分析測試學報),2014,33(11):1219-1223.
[4]Liu Y Q,Chen Q,Yu H,Tang H H.J.Instrum.Anal.(劉永強,陳倩,于泓,唐慧慧.分析測試學報),2014,33(10):1154-1159.
[5]Palmano K P,Elgarb D F.J.Chromatogr.A,2002,947:307 -311.
[6]Elgar D F,Norris C S,Ayers J S,Pritchard M,Otter D E,Palmano K P.J.Chromatogr.A,2000,878:183 -196.
[7]Silva R B,Collins K E,Collins C H.J.Chromatogr.A,2000,869:137-141.
[8]Kirklanda J J,Hendersona J W,Destefanoa J J,van Straten M A,Claessens H A.J.Chromatogr.A,1997,762:97 -112.
[9]Huck C W,Bakry R,Bonn G K.Eng.Life Sci.,2005,5(5):431 -435.
[10]Huck C W,Bonn G K.Chem.Eng.Technol.,2005,28(12):1457 -1472.
[11]Li P.Study on the Manufature of Nano-films.Chongqing:Chongqing University(李鵬.納米薄膜材料制備工藝研究.重慶:重慶大學),2004.
[12]Tian Z W.Preparation and Adsorptive Property of Hydrogen-bonded Hypercrosslinked Adsorbents Modified by Difunctional Groups.Zhenjiang:Jiangsu University(田志偉.雙官能團修飾氫鍵型超高交聯吸附樹脂的合成及其吸附性能研究.鎮江:江蘇大學),2011.
[13]Zhang Y,Huang Y M,Carr P W.J.Sep.Sci.,2011,34:1407-1422.
[14]Thompson J D,Carr P W.Anal.Chem.,2002,74(16):4150-4159.
[15]Yan B W,Zhao J H,Brown J S,Blackwell J,Carr P W.Anal.Chem.,2000,72(6):1253-1262.
[16]Heinisch S,Rocca J L.J.Chromatogr.A,2009,1216:642-658.
