木質素胺/聚酰胺-環氧樹脂體系的固化動力學研究
2015-01-06 05:22:07關麗珠邸明偉
粘接 2015年6期
關麗珠,邸明偉
(東北林業大學生物質材料科學與技術教育部重點實驗室,黑龍江 哈爾濱 150040)
木質素胺/聚酰胺-環氧樹脂體系的固化動力學研究
關麗珠,邸明偉
(東北林業大學生物質材料科學與技術教育部重點實驗室,黑龍江 哈爾濱 150040)
將玉米秸稈酶解木質素胺化改性后制得木質素胺,使用非等溫DSC掃描方法對木質素胺/聚酰胺-環氧樹脂體系的固化反應動力學進行了研究。分別通過Crane的n級反應法和Málek的最大概然機理函數法確定了固化體系的反應機理函數,計算了固化反應的動力學參數值,擬合了固化反應動力學模型,并通過外推法優化了固化工藝。結果表明,Málek的自催化反應模型在5~20 K/min的升溫速率下與實驗數值誤差較小,求解的模型更適合描述該體系的固化反應過程;對于木質素胺/聚酰胺的固化體系來說,優選的固化工藝為90 ℃/2 h+130 ℃/1 h。
環氧樹脂;聚酰胺;木質素胺;非等溫DSC;固化動力學
1 前言
環氧樹脂以其優良的粘接、耐腐蝕、絕緣和加工性能,廣泛應用于交通運輸、航空航天、建筑、包裝、水利電力等領域,已成為各工業領域不可缺少的基礎材料[1]。聚酰胺可以常溫固化環氧樹脂,同時又具有一定的增韌作用,因而已成為環氧樹脂的重要固化劑之一[2]。單一的聚酰胺固化劑固化的環氧樹脂耐熱性不高,通過添加其他含有苯環類的胺類固化劑,可以改善其耐熱性[3~5]。酶解木質素是秸稈酶解生物煉制過程中的副產物。作為多酚類的芳基化合物,酶解木質素具有較高的化學反應活性,同時又具有非水溶性,與聚合物相容性較好,因而可用于聚合物的改性[6~9]。研究表明,酶解木質素可以參與環氧樹脂的固化,從而改進環氧樹脂的耐熱性[10,11]。而作為酶解木質素的胺化產物,帶有活潑氫的木質素胺更易于參與環氧樹脂的固化,從而提高環氧樹脂的耐熱性能。本文采用Mannich反應以玉米秸稈酶解木質素和二乙烯三胺制備了木質素胺,將木質素胺與聚酰胺混合用于固化環氧樹脂。采用非等溫差示掃描量熱法(DSC)研究了木質素胺/聚酰胺-環氧樹脂體系的固化反應動力學,優化了固化工藝。
2 實驗部分
2.1 實驗原料
玉米秸稈酶解木質素,吉林松原來禾化學有限公司;木質素胺,實驗室自制,N含量68.6 mg/g;雙酚A型環氧樹脂E-51,中國南通星辰合成材料有限公司;聚酰胺TY-200,天津燕海化學有限公司。
2.2 DSC測試
按照前期實驗結果,選取木質素胺/聚酰胺的質量比為15:85。室溫下將木質素胺與聚酰胺混合均勻后,再與環氧樹脂混合,待用。其中聚酰胺與環氧樹脂的質量比為1:1。采用德國NETZSCH公司的DSC 204F1型差示掃描量熱儀進行不同升溫速率(5、10、15、20 K/min)的DSC掃描。每次測試試樣約10 mg左右,掃描溫度范圍為25~250 ℃。
3 反應模型的確定
DSC測量的是樣品與參比之間的熱流速率與時間或溫度的關系,應用模型擬合法對環氧樹脂固化行為進行動力學分析應基于以下3個基本假設[12]:
(1)假設固化速率與熱流速率成正比,即式(1):

(2)假設動力學分析的基本速率方程為式(2):
(3)假設固化反應的放熱焓與固化度成正比。
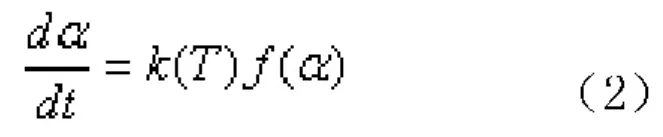
若是n級反應,則反應速率方程中的函數dα/dt=k(T)(1-α)n。若是自催化反應,根據Málek提出的判斷標準[13],反應體系可以用?esták等人[14]對非等溫條件下的固相反應進行研究所提出的雙參數模型:dα/dt=k(T)αm(1-α)n來表示,其中α為固化度,m、n為反應級數。
3.1 n級反應模型參數的求取
3.1.1 反應活化能和頻率因子的確定
固化反應動力學參數的確定對于研究體系的固化反應有著重要的作用。固化反應能否進行取決于表觀活化能Ea的 大小,當反應體系積累的能量大于表觀活化能Ea時 ,反應才能進行。而固化體系的反應復雜程度則取決于n,通過Ea、 n的計算可粗略推算固化反應的機理。
Kissinger[15]方程恰好符合對不同升溫速率下的DSC譜圖進行動力學處理。它假設在固化反應體系中,最大速率發生在固化反應放熱峰的峰頂溫度,反應級數n在固化過程中保持不變,適用于n級反應模型[16]假設的計算。
由動力學方程、Arrhenius方程和F(α)=(1-α)n可以得到式(3):
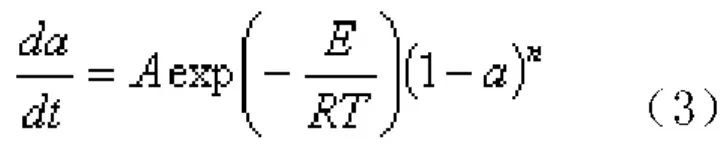
式(3)兩邊微分,當T=TP(Tp為 DSC峰頂溫度)時, ,Kissinger認為,與升溫速率β無關,其值近似等于1,因此Kissinger方程變為式(4):
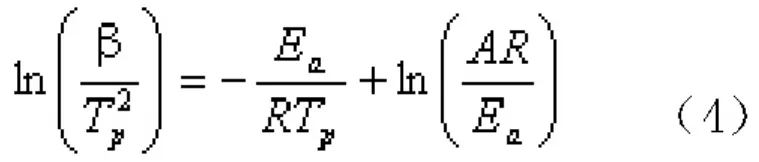
3.1.2 反應級數的確定
采用Crane[17]方程,對動力學參數進行線性擬合可以計算出n。由于Ea/(nR)>>2TP,因此Crane方程可以簡化為式(5):
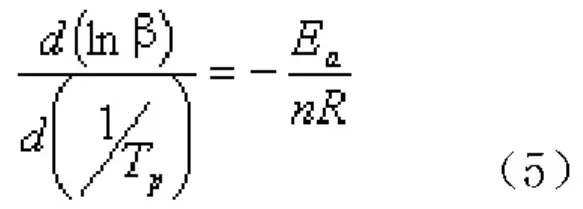
Ozawa等[18,19]提出了等轉化率積分法,在非等溫條件下作多組DSC實驗的lnβ-1/T圖,等溫條件下則作多組DSC實驗的lnt-1/T圖,對所得圖形進行線性回歸,斜率即為不同固化度時的活化能。
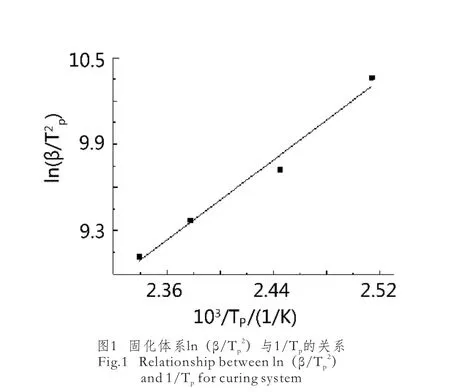
以lnβ對103/Tp作圖,得到一條直線,從直線斜率求解n。如圖2所示。
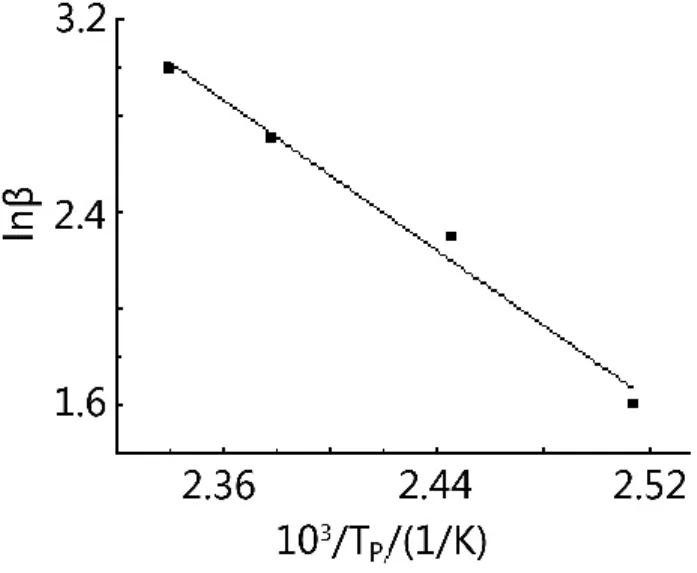
圖2 固化體系lnβ與1/Tp的 關系Fig.2 Relationship between lnβ and 1/Tpfor curing system
由式(5)及圖2可以得到線性方程:lnβ=-7.7634/Tp+21.185,R2=0.9864,由此方程計算得到n=0.89,說明該環氧樹脂體系的固化反應為復雜反應。
3.2 自催化反應模型參數的求取
Málek法是由定義函數y(α)、Z(α)來確定反應激勵函數f(α)的一種方法。此方法是通過分析不同升溫速率5、10、15、20K/min的固化參數,利用最大概然函數機理,求取Ea、 A、m、n動力學參數值,進而模擬得到固化機理函數。
3.2.1 活化能Ea的求取
首先將式(5)兩邊取對數得到:

通過Ozawa[18,19]提出的等轉化率積分法,分別將4個升溫速率下的固化度α=0.1、0. 2……0.9所對應的ln(dα/dt)、1/T的數值進行線性擬合,得到直線的斜率Ea/R,即可求出該固化度下對應的活化能Ea, 其中f(α)的形式對Ea的 數值沒有影響。然后求解平均值即可得到反應體系的Ea平 均值為52.11 kJ/mol。
由求取的活化能與轉化率的關系可以看出,當固化度α在0.1~0.5的時候,活化能的變化隨著固化度的變化較小;當固化度大于0.5后,體系活化能降低,且與平均值偏差較大。因為隨著反應的進行,體系溫度的升高使分子得到能量,從常態變化為容易發生反應的活躍狀態,從而使反應容易進行,活化能降低。
3.2.2 反應級數m、n、A的求取
為了準確選擇符合實際固化過程的模型,Málek[13]引入了2個特殊方程式(7)和式(8):
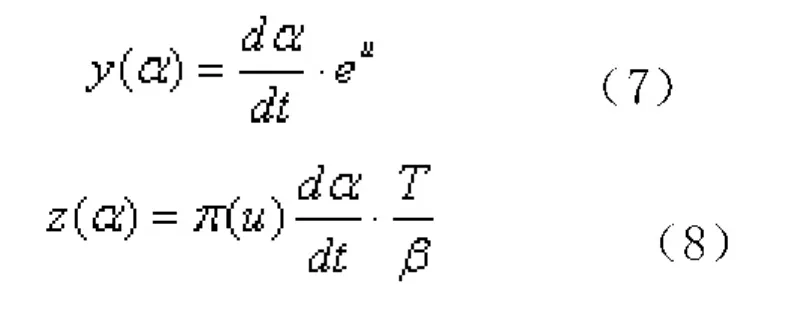
其中:π(u)為溫度的積分形式,使用Senum-Yang近似表達式(9)計算:
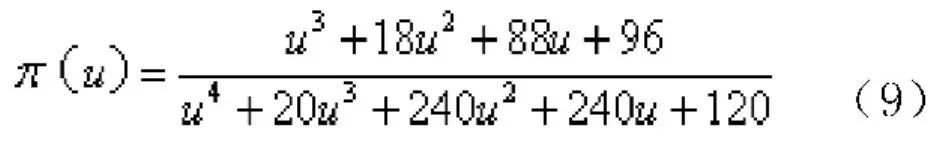
反應速率方程f(α)正比于y(α)和z(α),可以通過對y(α)和z(α)的曲線來判斷速率方程f(α)的變化趨勢,對y(α)和z(α)的頂點和形狀進行分析,即可求出合適的模型以描述固化過程。
由計算出的活化能Ea帶入式(7)和(8),分別求得不同升溫速率下y(α)和z(α)隨固化度變化的數值,然后將其在(0~1)范圍內標準化作圖3,y(α)和z(α)最大值對應的固化度分別標記為αM和。

圖3 函數y(α)和z(α)與轉化率α的關系曲線Fig.3 Relationship curves of function y(α) and z(α) versus conversion rate α
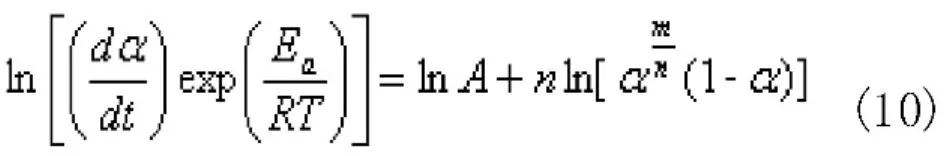
將DSC實驗數據帶入,將ln[(dα/dt)exp(Ea/RT)]對ln[αm/n(1-α)]作圖,擬合所得直線斜率即為n、截距為lnA,如圖4所示。令(10)式中m/n=p=αm/(1-αm) ,由此可以計算得到m。
表1為不同升溫速率下的自催化反應模型參數計算過程中物理量的計算值。
3.3 固化反應動力學模型
將上述2種方法求取的動力學參數帶入反應機理函數f(α),則得到木質素胺/聚酰胺-環氧樹脂固化體系的固化反應動力學模型:
n級反應模型式(11):

自催化反應模型式(12):
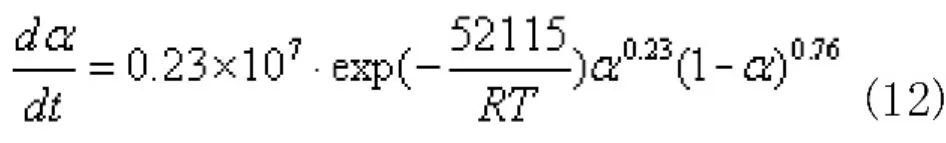
將2種反應模型的計算值與實驗值相比較,如圖5、圖6所示。
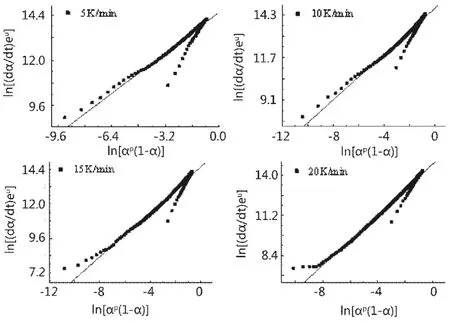
圖4 不同升溫速率下的ln[(dα/dt)eu]與ln[αp(1-α)]的關系曲線Fig.4 Relationship curves of ln[(dα/dt)eu] versus ln[αp(1-α)] under various heating rate

表1 不同升溫速率下的計算參數Tab.1 Curing kinetic parameters under various heating rate

圖5 n級反應模型的計算值與實驗值比較Fig.5 Comparison of calculated and measured values for n-order reaction model
由圖5、6可以看出,由n級反應模型求取的固化動力學反應模型在每一種升溫速率下都與實驗值有一定的誤差,而由Málek最大概然函數法求取的固化動力學模型在升溫速率為5 K/min和15 K/min的時候基本與實驗值相符,而在10 K/min、20 K/min的時候雖有一定的誤差,但也比n級方法求取的計算值與實驗值誤差小。因此相對來說自催化固化反應模型比n級反應固化動力學模型更加適合模擬木質素胺/聚酰胺/環氧樹脂體系的固化反應過程。
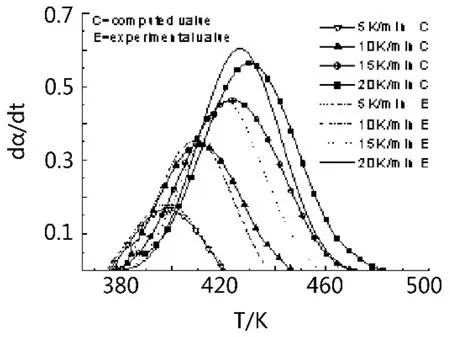
圖6 自催化反應模型的計算值與實驗值比較Fig.6 Comparison of calculated and measured values for autocatalytic reaction model
3.4 體系固化工藝的確定
以放熱峰Ti、Tp和Tf對 升溫速率β作圖,線性擬合后可以外推得到β=0 K/min對應的溫度值。此3點溫度可以分別代表凝膠溫度Tgel、
固化溫度Tcure和后處理溫度Ttreat, 通過此方法得到的Tgel、Tcure以及Ttreat分 別為89.15 ℃、115.35 ℃和144 ℃。為了保證固化完全,通常后固化溫度選在Tcure和Treat之間[20],為此本文的固化體系可以選在90 ℃固化一段時間后再升至130 ℃左右進行后固化,即固化工藝可為90 ℃/2 h+130 ℃/1 h,測得其固化度為96.5%,說明固化反應基本完全。
4 結論
木質素胺可與聚酰胺混合用于固化環氧樹脂,對于木質素胺/聚酰胺-環氧樹脂混合體系來說,Málek的自催化反應模型更適合描述該體系的固化反應過程。利用外推法得到Tgel、Tcure以及Ttreat分 別為89.15 ℃、115.35℃和144 ℃,優選的固化工藝為90 ℃/2 h+130 ℃/1 h。

[1]陳凈. 國內環氧樹脂行業現狀及未來發展趨勢[J]. 中國涂料, 2012, 27(7):25-27.
[2]夏建陵, 孫小梅, 王定選. 環氧樹脂聚酰胺網絡體系性能研究[J]. 熱固性樹脂, 2005, 20(6):14-17.
[3]董新, 蔡智奇, 皮丕輝, 等. 環氧樹脂/聚酰胺/DDM體系的固化行為及力學性能[J]. 熱固性樹脂, 2011,26(1):21-24.
[4]胡高平, 肖衛東. 酚醛胺-低分子聚酰胺協同固化環氧樹脂的研究[J]. 熱固性樹脂, 2002, 17(2):25-26.
[5]梁鳳飛, 陳立新, 李凡. 間苯二胺/低分子聚酰胺協同固化EP膠粘劑的研究[J]. 中國膠黏劑, 2011, 20(12):5-8.
[6]Stewart D. Lignin as a base material for materials applications: Chemistry, application and economics[J]. Industrial Crops and Products, 2008, 27(2):202-207.
[7]陳云平, 程賢甦. 木質素基復合材料的制備及在乙丙橡膠中的應用[J]. 現代化工, 2009 (2):36-38.
[8]陳為健, 程賢甦, 方潤. 木質素基聚酯型環氧樹脂的制備及表征[J]. 纖維素科學與技術, 2009,17(2):1-5.
[9]Park Y, Doherty W O S, Halley P J. Developing lignin-based resin coatings and composites[J]. Industrial Crops and Products, 2008,27(2):163-167.
[10]Quanfu Yin, Weijun Yang, Chengjun Sun, et al. Preparation and properties of lignin-epoxy resincomposite[J]. Bio Resources, 2012,7(4):5737-5748.
[11]Xianzhi Kong, Zhifeng Xu, Lizhu Guan, et al. Study on polyblending epoxy resin adhesive with lignin I-curing temperature[J]. International Journal of Adhesion & Adhesives, 2014,48:75-79.
[12]Vyazovkin S.Thermal analysis[J]. Analytical Chemistry,2008,80(12):4301-4316.
[13]Má lek J. The kinetic analysis of nonisothermal data[J]. Thermochimica Acta, 1992, 200:257-269.
[14]?esták J, Berggren G. Study of the kinetics of the mechanism of solid-state reactions at increasing temperatures[J]. Thermochimica Acta, 1971, 3(1):1-12.
[15]Kissinger H E. Reaction kinetics in differential thermal analysis[J]. Analytical Chemistry, 1957, 29(11): 1702-1706.
[16]Macan J, Brnardi? I, Ivankovi? M, et al. DSC study of cure kinetics of DGEBA-based epoxy resin with poly(oxypropylene) diamine[J]. Journal of Thermal Analysis and Calorimetry,2005, 81(2):369-373.
[17]Crane L W, Dynes P J, Kaelble D H. Analysis of curing kinetics in polymer composites[J]. Polymer Letters Edition, 1973, 11(8):533-540.
[18]Ozawa T. Kinetic analysis of derivative curves in thermal analysis[J]. Journal of Thermal Analysis and Calorimetry, 1970,2(3):301-324.
[19]Ozawa T.Kinetics of non-isothermal crystallization[J].Polymer,1971,12(3):150-158
[20]代曉青, 曾竟成, 劉鈞, 等. RFI用環氧樹脂固化工藝研究[J]. 國防科技大學學報. 2007,29(3):22-26.
Study on curing kinetics of lignin amine/polyamine-epoxy resin blends
GUAN Li-zhu, DI Ming-wei
(Key Laboratory of Bio-Based Material Science&Technology (Ministry of Education), Northeast Forestry University, Harbin, Heilongjiang 150040,China)
In this study, the lignin obtained from enzymically hydrolyzed corn stalk was used to synthesize the lignin amine via Mannich reaction. Using the non-isothermal differential scanning calorinetry (DSC) analysis the curing kinetics of lignin amine/polyamine-epoxy resin blends was investigated, and using the n-order reaction method of Crane and the most probable mechanism functions of Málek the reaction mechanism function of curing system was determined. Simultaneously, the curing kinetic parameters were calculated and the cure kinetic model was fitted. And the curing processing was optimized by extrapolation method. The results showed that the autocatalytic reaction model of Málek was close to the experiment data under the heating rate of 5-20 K/min and more suitable to characterize the cure reaction procedure. For the mixed curing system of lignin amine and polyamine, the optimized curing processing was determined as 90℃ for 2 h, and then l30℃ for 1 h.
epoxy resin;polyamine;lignin amine;non-isothermal DSC;curing kinetics
TQ433.4+37
A
1001-5922(2015)06-0031-06
2014-09-18
關麗珠(1989-),女,在讀碩士研究生。
邸明偉(1972-),男,教授,博導,主要研究方向為生物質復合材料及膠粘劑。E-mail:dimingwei@126.com。
黑龍江省科學基金資助項目(C201335、A201205)、黑龍江省哈爾濱市科技創新人才研究專項資金項目(2014RFXXJ066)。
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
中學生數理化·七年級數學人教版(2020年10期)2020-11-26 08:24:50
數學物理學報(2020年2期)2020-06-02 11:29:24
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
光學精密工程(2016年6期)2016-11-07 09:07:19
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
新高考·高一物理(2014年1期)2014-09-18 01:26:07
