椎管內原始神經外胚層腫瘤1例報告
2015-02-07 12:10:13孟小童賀石生
中國醫科大學學報 2015年8期
孟小童,賀石生
(同濟大學附屬上海市第十人民醫院脊柱外科,上海 200072)
·短篇論著·
椎管內原始神經外胚層腫瘤1例報告
Primitive NeurotodermalTumour in SpinalCanal:ACase Reportand Literature Review
孟小童,賀石生
(同濟大學附屬上海市第十人民醫院脊柱外科,上海 200072)
原始神經外胚層腫瘤(PNET)是主要發生于兒童與青年人群且組織來源尚未明確的一類罕見惡性腫瘤。我們報告1例以下腰痛及右下肢麻木就醫的中年女性PNET,該患者在我院行椎管內腫瘤切除術合并化療。初步治療結束后,原有的脊髓壓迫癥狀緩解,隨訪4個月,因腫瘤復發死亡。本文將討論PNET的臨床表現、鑒別診斷、治療方案(包含外科切除與局部放療結合)等。
原始神經外胚層腫瘤;脊柱腫瘤;化學治療;中年女性
原始神經外胚層腫瘤(primitive neurotodermal tumour,PNET)是一類極罕見,高侵蝕性、不明組織來源且多發于青年男性的惡性腫瘤,由Hart和Earle1973年首次報道[1],迄今為止,在各類文獻報告不多于100例。本例PNET發生于骨外而且膨大進入腰椎管,從而引起了脊髓壓迫癥狀。以往只有少數類病例被報道[2~4]。
1 臨床資料
患者,女,60歲,因“腰痛伴右下肢麻木1年”入院。入院查體:血壓130/80 mmHg,體溫36.0~36.7℃,步態不穩,腰椎活動稍受限,右下肢L2、L3支配區域感覺減退,肌力Ⅲ級,右下肢膝反射、踝反射減弱,雙Hoffmann征(-)。CT下可見L2、L3間大小邊界不清的腫塊,該腫塊在增強MRI上呈均勻增強,TI、T2呈一致信號(圖1)腹部和骨盆CT、X線胸片均未見異常,無腹水征象,無腹膜后或淋巴結大范圍轉移征象。
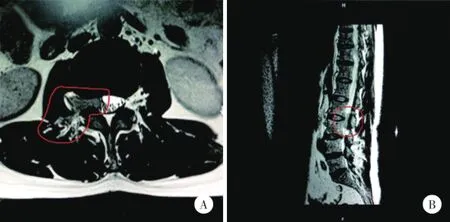
圖1 患者腰椎增強磁共振平掃結果
免疫組織化學檢查表明該惡性腫瘤細胞膜上有CD99表達。免疫組織化學標記:Ki-67(+)、VI(++)、S-100(+)、NeuN(+)、CD99(++)、NBT(-)、NSE(+)、CD7(-)、TdT(-)、De(-)、CAM5.2(-)、EMA(-)、SCLC(-)、Chr(-)、SMA(-)、HMB45(-),組織病理學檢查可見原始神經外胚層腫瘤的瘤細胞呈小圓形或卵圓形,深染,染色質細,核仁不清,癌細胞呈實性、片狀、分葉狀、腺泡狀或索條狀排列,其間有豐富的纖維和血管分隔;腫瘤細胞圍繞血管周圍呈假菊花團狀,中心為絲狀纖維,無空腔、血管或瘤細胞的壞死物,稱之為Homer-Wright菊形團(圖2)。
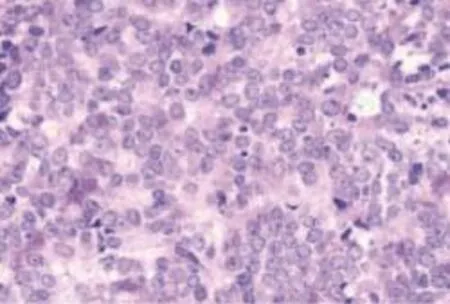
圖2 患者術中切除組織行免疫組化染色 ×40
行椎管內腫物切除術,當L2、L3椎體的半椎板均被切除后,1個4.9 cm×2.1 cm×1.8 cm、邊界清楚的組織團塊出現在視野中,該團塊與右側L2神經根毗鄰,并且通過L2、L3椎體間的椎間孔向椎管內膨出。術中探及L3神經根活動度佳,且所有的切緣均未見與腫瘤組織明顯粘連。光鏡下,切除的組織團塊的切面呈白色、不規則、實性,且有許多小凸起。術后,該病人腰痛癥狀緩解,麻木稍有存在,Nurick分級為1,無特殊不適主訴,術后立即轉至外院行放射性治療,4個月后因全身多臟器轉移造成的多器官衰竭死亡。
2 討論
原始神經外胚層腫瘤的年發病率大約在每十萬人0.2至0.4人[5]。該病常表現為迅速增大的,伴有疼痛的軟組織腫塊,且常引起局部壓迫癥狀。原始神經外胚層腫瘤已被定性為高度惡性、高侵襲性、高轉移性、高復發性、難診治性的惡性腫瘤,5年生存率為30%~40%[6],該數據在過去30年間沒有顯著改善[7]。
原始神經外胚層腫瘤被分為中樞性原始神經外胚層腫瘤(cPNET)和外周性原始神經外胚層腫瘤(pPNET)兩類,二者預后均較差。外周性原始神經外胚層腫瘤代表著發生于顱外且具有相同形態學和細胞學特性的惡性腫瘤細胞,包括發生于骨內或骨外的尤文氏肉瘤,發生于椎旁、腹腔內、腹膜后軟組織的外周性原始神經外胚層腫瘤以及發生于胸壁的Askin肉瘤[9~11]。外周性原始神經外胚層腫瘤與尤文氏肉瘤因在組織學檢查中均可見到小圓細胞而被認為是密切聯系的兩種惡性腫瘤,二者免疫組織化學檢測中,由微絲蛋白2(MIC2)基因編碼的糖類蛋白p30/32(CD99)均為強陽性,由于上述的免疫組織化學證據,超微結構,分子生物學相似性,近年來外周性原始神經外胚層腫瘤與尤文氏肉瘤被統一歸類于尤文家族,二者共享相同的染色體易位位點,而二者的主要區別在于二者之間具有不同的分化程度。
以往類似的病例因腫瘤常與硬膜毗連而常常被診斷為星形細胞瘤,室管膜瘤或神經纖維瘤。由于腫物通過椎間孔膨出至腰椎管內,故在診斷之初,我們曾一度懷疑本文的病例為室管膜瘤,然而室管膜瘤經完全切除,預后是很好的。
外周性原始神經外胚層腫瘤是一種診斷較為困難的高侵襲性癌,目前尚無有效治療手段,外科切除術若不能完全切除腫瘤則會造成90%以上的患者出現術后復發。
[1]Hart M,Eade KM.Primitive neuroeetodermal tumors of the brain in children[J].Cancer,1973,32:890-897.
[2]Virani M,Jain S.Primary intraspinal primitive neuroectodermal tumor(PNET):a rare occurrence[J].Neurol India,2002,50(1):75-80.
[3]Nayak P,Rao KM,Sahoo GC,et al.Primary thoracic primitive neuroectodermal tumor mimicking as neurofibroma[J].J Neurol India,2011,59(4):648-649.
[4]Patnaik A,Mishra SS.Review of spinal neuroectodermal tumor[J]. Brit J Neurosurg,2013,27(1):2-6.
[5]Valle J,Eatock M,Clueit B.A systematic review of non-surgical treatments for pancreatic neuroendocrine tumors[J].Cancer Treat Rev,2014,40(3):376-389.
[6]Pape UF,Bohmig M,Berndt U,et al.Survival and clinical outcome of patients with neuroendocrine tumors of the gastroenteropancreatic tract in a German referral center[J].Ann N Y Acad Sci,2004,1014:222-233.
[7]Marinsek ZP,Kavalar R,Jereb B.Ewing sarcoma/PNET:27 years of experience in Slovenia[J].Pediatr Hematol Oneol,2006,23(4):355-367.
[8]Kampman WA,Kros JM,De Jong TH,et al.Primitive neuroectodermal tumours(PNETs)located in the spinal canal;the relevance of classification as central or peripheral PNET:case report of a primary spinal PNET occurrence with a critical literature review[J].Neuro-oncology,2006,77(1):65-72.
[9]Khong P,Chan GC,Shek TW.Imaging of peripheral PNET:common and uncommon locations[J].Clin Radiol,2002,57(4):272-277.
[10]Dehner L.Peripheral and central primitive neuroectodermal tumors z anosologic concept seeking a consensus[J].Arch Pathol Lab Med,1986,110(11):997-1005.
[11]Winer-Muram HT,Kauffman WM,Gronemeyer SA.Primitive neuroectodermal tumors of the chest wall(Askin tumors):CT and MR findings[J].AJR.Am J Obstet Gynecol,1993,161(2):265-268.
(編輯 裘孝琦)
R739.42
A
0258-4646(2015)08-0764-02
孟小童(1988-),男,碩士研究生.
賀石生,E-mail:mxtsss0120@aliyun.com
2014-11-15
網絡出版時間:
