潔凈瓶胚料在塑瓶大輸液生產中回收利用的研究
2015-03-15 02:03:07崔良峰黃桂華
藥學研究 2015年8期
崔良峰,黃桂華
(山東大學藥學院,山東濟南250012)
·工業藥學·
潔凈瓶胚料在塑瓶大輸液生產中回收利用的研究
崔良峰,黃桂華
(山東大學藥學院,山東濟南250012)
目的將塑瓶大輸液生產中灌裝藥液前產生的潔凈瓶、胚,重新粉碎成一定粒徑的顆粒后,與新PP粒料按一定比例混合后,重新吹制成PP輸液瓶,用于PP塑瓶大輸液產品的生產。通過對回收利用過程中的各種風險和影響因素進行識別,采取針對性的控制措施,確保最終產品質量不受影響。方法①進行風險識別,確定風險點并據此進行風險控制與工藝研究;②選取3個不同廠家的PP粒料,進行不同混合比例的制瓶研究;③將不同混合比例的PP瓶分別用于0.9%氯化鈉注射液、5%葡萄糖注射液的生產,對成品質量檢驗并進行加速試驗考察與包材相容性研究,確定使用混合料對制劑產品質量與原工藝生產的制劑產品質量是否存在差異。結果及結論同質潔凈瓶胚料按照合理的比例進行回收利用,其制劑產品的質量,通過注射劑與PP包裝容器的相容性研究和穩定性考察數據與正常生產的制劑產品對比,數據無明顯的差別,同時進行生物實驗均符合要求,能夠保證產品質量安全、穩定。
風險識別;PP粒料;加速穩定性考察;包材相容性
PP塑瓶已成為目前國內臨床應用最廣的輸液包裝形式,市場占有率已達60%以上,年產量80億瓶袋。目前國內近80家塑瓶輸液生產廠家,通常使用國產設備采用注-吹生產工藝進行制瓶,制瓶合格率在97%左右,因此會產生大量的瓶、胚廢品,造成資源的浪費,且不利于環境保護[1~3]。初步統計,造成直接經濟損失近5 000萬元。根據《藥品包裝使用手冊》第六章之“制瓶主要生產工藝”,對加回料“應使用同質潔凈回料,并保持適當和穩定的比例”的要求[1],為更好的利用資源,我們對塑瓶大輸液生產中灌裝藥液前產生的,僅用過1次的潔凈PP瓶、胚,統一收集粉碎后,作為同質潔凈瓶胚料。再通過混合機與正常使用的新PP料按一定比例進行均勻混合,重新制作瓶胚吹制成PP輸液瓶,用于PP塑瓶大輸液的生產。本文針對同質潔凈瓶胚料在塑瓶大輸液生產中進行回收利用的工藝研究進行試驗和討論。
1 風險識別及風險點的控制
基于聚丙烯PP粒料為常用注塑材料,其化學性質穩定,耐熱性能較好,耐彎曲疲勞性強,長時間加熱至300℃以上方可發生降解[2]。使用注-吹工藝生產的聚丙烯輸液瓶,生產過程中的受熱溫度低于300℃,且受熱時間相對較短,未發生顏色及化學性質的變化。同質潔凈瓶胚料的使用是將清潔、未污染瓶胚、廢瓶經過粉碎機進行粉碎為顆粒后與同廠家粒料混合使用,整個過程只是物理性質的改變,不發生相應的化學變化。
1.1 塑瓶清潔同質回料使用會存在潛在的風險 根據藥品生產質量管理規范(2010年修訂)質量風險管理第十四條“應當根據科學知識及經驗對質量風險進行評估,以保證產品質量”[3]。因此,采用“魚骨圖”的風險分析模式進行風險識別,見圖1。

圖3 塑瓶清潔同質回料使用存在風險魚骨分析圖
1.2 分析與討論 通過風險識別,確定影響聚丙烯輸液瓶質量的關鍵因素為:注塑吹瓶設備運行參數、回收料的潔凈度、粉碎的粒徑和均勻度、與新粒料的混合比例、重復回用的次數;對輸液產品質量的影響主要為:聚丙烯輸液瓶質量檢測、PP制劑質量、穩定性考察結果、藥液相容性等,因此結合關鍵影響因素進行工藝研究,可確保產品質量的安全、穩定與可靠。
2 材料、設備與研究方法
2.1 材料與設備
2.1.1 PP粒料 李長榮化學工業股份有限公司、韓國曉星株式會社、韓國LG 3個廠家的粒料與同質潔凈瓶胚料。
2.1.2 生產設備與檢驗儀器 粉碎機、立式混合機,PP注塑機、OPP大輸液吹瓶機、灌裝機、滅菌器、高效液相色譜儀、微粒分析儀、酸度計、SBJ-6000塑瓶試驗機、IRPrestige-21紅外分光光度計、UV-2550紫外分光光度計、Gc-2010A高效氣相色譜儀等。
2.2 方法
2.2.1 不同混合比例粒料的制瓶生產 3種廠家粒料生產過程中產生的潔凈瓶胚料,粉碎為小于Φ10 mm的碎料后,分別按照10%、20%、50%的比例與同廠家的新PP粒料混合,按照聚丙烯輸液瓶生產工藝進行注-吹生產,吹制100、250、500 mL輸液瓶。制胚、制瓶工藝參數在原工藝參數的范圍內進行調整。制胚質量檢查為外觀、重量2個項目,制瓶質量檢查為外觀、重量、均勻度3個項目。
2.2.1.1 制胚 不同混料比例的粒料,在料斗中的流動性與純料比較無明顯差別,按照生產同規格純料瓶胚的注塑控制參數范圍進行數值設定,僅需參數微調,均可制得相應規格的瓶胚。
隨機抽取純料、10%、20%、50%混合料所制得不同規格的瓶胚,進行外觀和重量的比對。結果顯示,4種PP料所制得瓶胚的外觀、重量均符合標準要求,且稱重數據均勻,證明不同比例混合PP料與純料的性質及使用過程中的差別很小,對瓶胚的質量影響不明顯,外觀、瓶胚重量指標符合使用要求。
2.2.1.2 制瓶 不同混料比例的粒料制得的瓶胚,保持加熱燈管層數、燈管溫度按照純料瓶胚的控制參數設置,進行吹瓶生產。參數微調,均可制得合格輸液瓶。
隨即抽取3種混料比例瓶胚制得的輸液瓶,按照PP聚丙烯輸液瓶質量標準進行檢查比對,外觀、均勻度和重量均符合標準,50%混料比例的成品率略低,結果見表1。
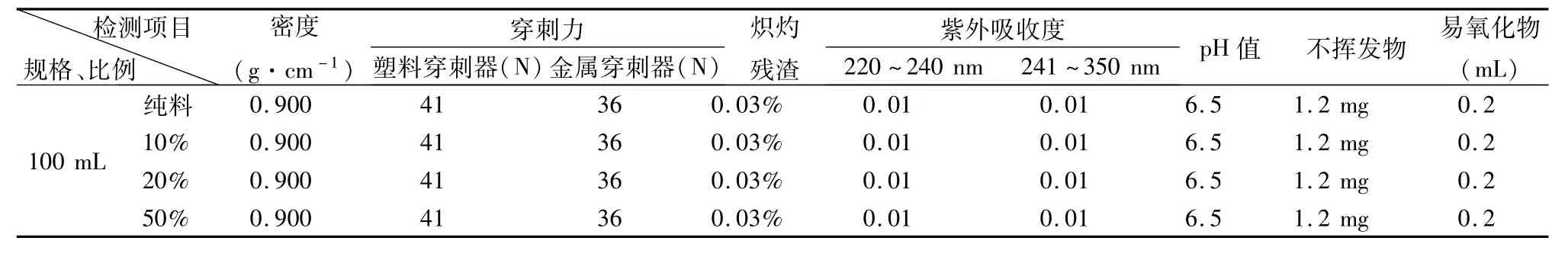
表1不同比例混料輸液瓶質量檢測數據匯總表
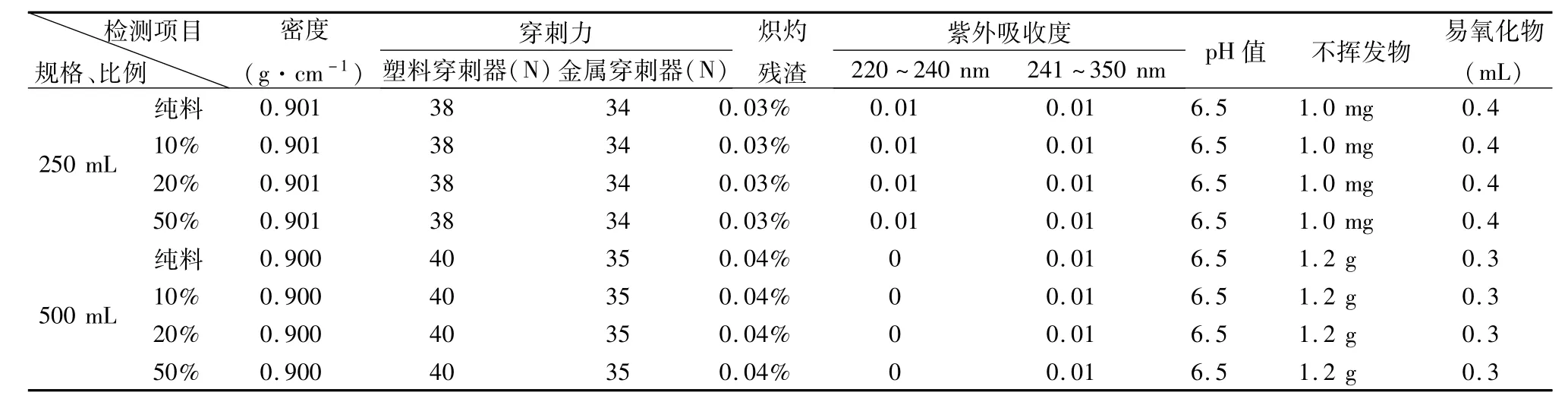
續表1
數據顯示,4種比例PP瓶質量對比無明顯差別,均符合聚丙烯輸液瓶質量YBB00022002標準。
2.2.2 混合料輸液瓶的制劑生產 按照PP塑瓶大容量注射劑生產工藝規程進行不同規格的0.9%氯化鈉注射液和5%葡萄糖注射液的灌封與滅菌。各取500瓶,對可見異物不溶性微粒、合格率情況進行數據統計,并對產品質量按標準進行全檢,應符合《中國藥典》2010年版質量規定,結果見表2。
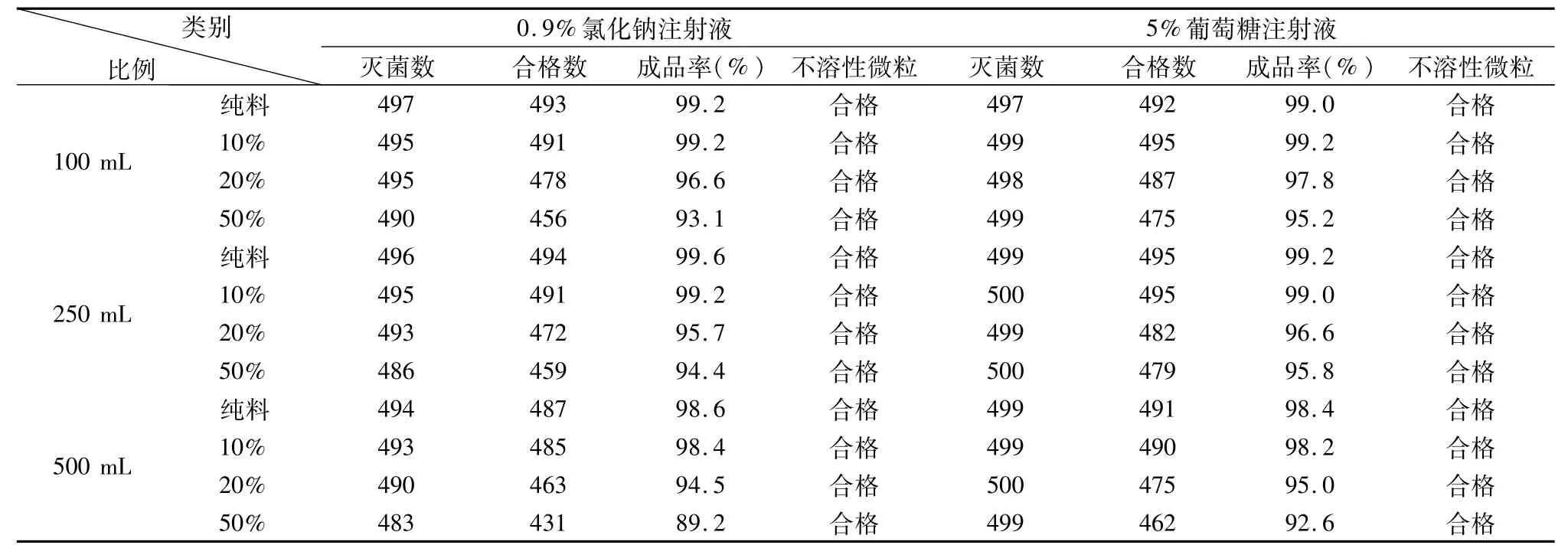
表2不同混料比例輸液產品檢測數據匯總表
2.2.3 加速實驗和穩定性考察 按照《中國藥典》2010年版(二部)附錄XIX C“原料藥與藥物制劑穩定性試驗指導原則”要求進行穩定性考察[4]。方法如下:
將上述注射液留樣樣品在溫度(40±2)℃、相對濕度20%±5%條件下放置6個月,分別于0、1、2、3、6個月末取樣檢查。
注射劑檢測項目:性狀、含量、pH值、可見異物、不溶性微粒、無菌、細菌內毒素、滲透壓摩爾濃度(0.9%氯化鈉注射液)、5-羥甲基糠醛(5%葡萄糖注射液)。其中無菌、細菌內毒素只在0月、6月末檢測。
PP輸液瓶分別在0、6月進行全檢,1、2、3月末取樣1次檢測溫度適應性、抗跌落、透明度、穿刺部位不滲透性、外觀、穿刺力、懸掛力。
2.2.3.1 聚丙烯瓶加速試驗 通過加速實驗的數據顯示,各項檢測指標在0、1、2、3、6個月均應符合《中國藥典》2010年版(二部)葡萄糖注射液質量標準[4]。
2.2.3.2 制劑產品加速試驗考察(以100 mL 5%葡萄糖注射液,20%混料比例產品為例) 通過對4個混料比例生產的不同規格的產品進行加速試驗考察檢測數據顯示:性狀、含量、pH值、5-羥甲基糠醛(5%葡萄糖注射液)、可見異物、不溶性微粒、無菌、細菌內毒素等檢測結果對比,無明顯差別,均符合5%葡萄糖注射液質量標準,結果見表3。
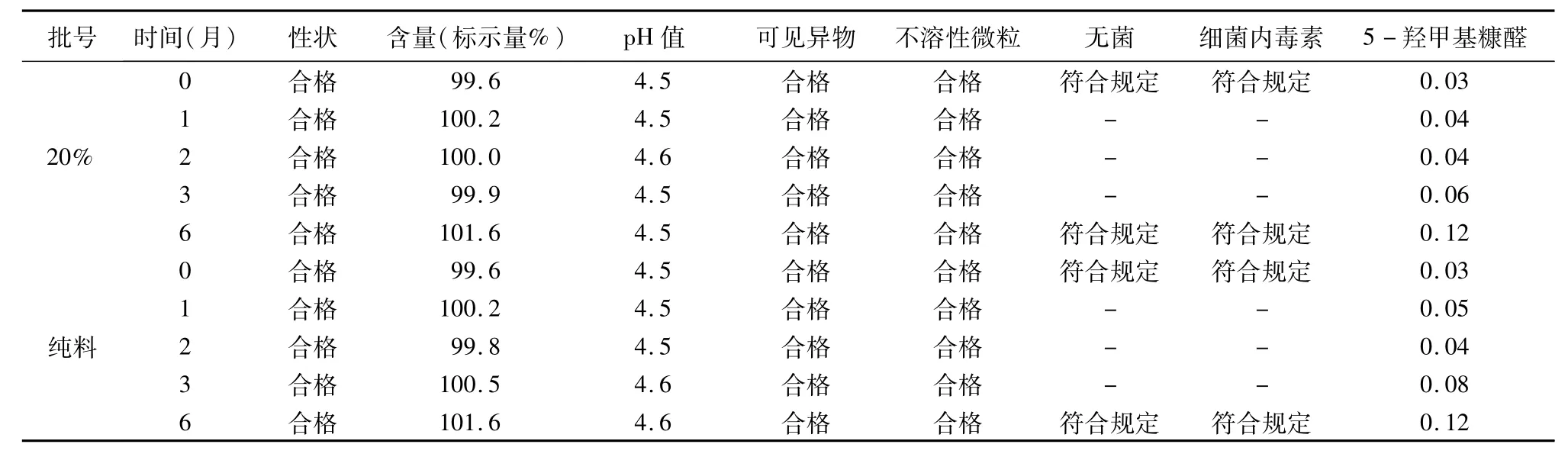
表3 制劑產品加速試驗考察表
2.2.4 包材相容性研究(遷移成分檢測) 藥品包裝材料與相容性試驗是指為考察藥品包裝材料與藥物之間是否發生遷移或吸附等現象,進而影響藥品質量的一種試驗。根據“化學藥品注射劑與塑料包裝材料相容性研究技術指導原則(試行)”[5]與“藥品包裝材料與藥物相容性試驗指導原則”規定,結合《歐洲藥典》第5版的方法重點進行遷移試驗與生物試驗。
2.2.4.1 樣品中的抗氧劑及其遷移實驗 參照《歐洲藥典》第5版,每種聚丙烯粒料各500 g,100、250、500 mL規格4個比例瓶胚、輸液瓶各100只,進行抗氧劑含量的檢查。對其瓶制劑產品加速考察0 d、6個月時取樣進行遷移試驗,檢測所含抗氧劑遷移情況,并對其檢測數據進行對比。
抗氧劑檢測實驗結果顯示:樣品中抗氧劑1010和抗氧劑330的單個含量分別為0.03%和0.02%,均小于0.05%標準值,總量均未超過0.1%。均符合《歐洲藥典》第5版3. 1.6(非腸道制劑及眼科制劑用容器用聚丙烯)規定的單個抗氧劑不得過0.3%,總量不得過0.3%的限度要求。
分別對115℃、40 min、115℃、30 min兩種滅菌條件下100、250、500mL規格4種混料比例生產的0.9%氯化鈉注射液和5%葡萄糖注射液進行的遷移實驗的結果顯示,抗氧劑均未檢出,未發現遷移。
2.2.4.3 鎂、鋁元素及其遷移試驗 《歐洲藥典》第5版“非腸道制劑及眼科制劑用容器用聚丙烯”中對水滑石提出了不多于0.5%的要求。相當于由水滑石引入的鎂不多于0. 12%,鋁不多于0.04%。通過試驗,不同規格、不同混料比例樣品中測得的鎂、鋁元素均小于0.01%,均小于由水滑石引入的鎂、鋁限值。
分別對115℃、40 min、115℃、30 min兩種滅菌條件下,100、250、500mL規格4種混料比例生產的0.9%氯化鈉注射液和5%葡萄糖注射液進行的遷移實驗的結果顯示,抗氧劑均未檢出,鎂、鋁元素在注射液中的遷移量均較小,均未超過0.05μg·mL-1。
2.2.4.4 材料中的苯乙烯單體殘留 檢測檢測數據顯示:100、250、500 mL規格4種混料比例聚丙烯輸液瓶樣品中的苯乙烯單體殘留量均小于0.6μg·g-1,符合規定要求。
2.2.5 動物實驗 依據國家藥品包裝容器(材料)方法標準(試行)YBB00032003的標準進行檢測。
將本次實驗所制得4種混料比例PP瓶制劑產品分別進行動物實驗項目檢測,評價產品質量的用藥安全性,結果見表4。

表4 不同比例混料輸液產品動物實驗檢測結果表
動物實驗數據顯示:動物實驗項目均符合國家藥品包裝容器(材料)方法標準(試行)。
3 結果及討論
目前的研究結果表明:按10%比例進行同質粒料的回收,在保持現有制胚、吹瓶工藝參數不變的情況下,能夠保證最佳合格率。進行0.9%氯化鈉注射液與5%葡萄糖注射液的生產,其PP輸液瓶質量及產品質量符合標準要求,加速穩定性考察、包材與藥液相容性研究各項指標均合格,且動物實驗的各項指標也符合國家標準要求,能夠保證產品的安全性。生產企業可依據研究數據,在生產車間進行0.9%氯化鈉注射液、5%葡萄糖注射液輸液產品的批量生產工藝驗證,對驗證產品進行穩定性考察,確認產品質量穩定。提報包材注冊變更,通過審批后可以進行規模化生產。
[1] 張英.國內塑料輸液包裝市場現狀及發展前景[J].石化技術,2011,18(1):63-66.
[2] 洪澤雄.淺析我國塑料包裝的發展新動向[J].輕工科技,2015,31(4):39-40.
[3] 徐興祥.做好中國塑料包裝減量化工作探討[J].上海包裝,2014,2:37-39.
[4] 中國醫藥包裝協會.藥品包裝實用手冊[M].北京:化學工業出版社,2003.
[5] 王華山.塑料注塑技術與實例[M].北京:化學工業出版社,2006.
[6] 國家食品藥品監督管理局.藥品生產質量管理規范(2010年修訂)[S].衛生部令第79號,2011-1-17.
[7] 國家藥典委員會.中華人民共和國藥典2010年版(二部)[S].北京:中國醫藥科技出版社,2010:附錄XIX,附錄199,928.
[8] 國家食品藥品監督管理局.化學藥品注射劑與塑料包裝材料相容性研究技術指導原則(試行)[S].國食藥監注[2012]267號,2012-9-7.
The research of clean bottle and bottle embryo recycle in the PP bottle infusion production
CUILiang-feng,HUANGGui-hua
(School of Pharmaceutical Sciences Shandong University,Jinan 250012,China)
ObjectiveCrush the clean plastic bottles and bottle embryos into a certain size of particles,add new pp aggregatemixed according to certain proportion,blow them into PP bottles again,the pp bottleswill be used in the production of pp bottle infusions.Through the identification of the risk and influence factor during the recycle of the pp materials,take corresponding controlmeasures to ensure the quality of the final products notbe affected.M ethods①Identify the risk,research in the risk controland production process according the risk point.②Select pp aggregate from 3 differentmanufacturers,research the different proportions ofmaking pp bottles.③Use the pp bottles in the production of0.9%sodium chloride injection and 5%glucose injection,test,research the accelerated stability study and the package material compatibility.Make sure if there is quality difference between the new productswhich usesmixing aggregate and original products.Resu lts and ConclusionRecycle the homogeneous clean plastic bottles and bottle embryos with a certain proportion.Research the accelerated stability study and the packagematerial compatibility,and compared with normal production,there is no obvious difference in the data,and the biologicalexperimentsmeet the requirementat the same time.Thismethod can ensure the product safety and stability.
Risk identification;PP aggregate;Accelerated stability study;Packagematerial compatibility
TQ460.4
A
2095-5375(2015)08-0490-004
崔良峰,男,工程師,研究方向:制藥工程,E-mail:qdyyclf1981@126.com
猜你喜歡
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
小學科學(學生版)(2020年10期)2020-10-28 07:52:12
中國化肥信息(2020年7期)2020-03-19 01:54:02
中國軍轉民(2017年6期)2018-01-31 02:22:28
海峽科技與產業(2016年3期)2016-05-17 04:32:12
汽車零部件(2014年11期)2014-09-18 11:57:16
