硼原子摻雜對碳納米管吸附Ne原子影響的第一性原理計算
2015-03-23 11:56:29溫述龍閆躍陽
原子與分子物理學報 2015年2期
溫述龍, 王 茜, 潘 敏, 閆躍陽, 黃 整
(1. 西南交通大學物理科學與技術學院, 成都 610031; 2. 中國工程物理研究院核物理與化學研究所, 綿陽 621900;3.西南交通大學超導與新能源研究開發中心, 成都 610031)
硼原子摻雜對碳納米管吸附Ne原子影響的第一性原理計算
溫述龍1, 王 茜2, 潘 敏3, 閆躍陽1, 黃 整1
(1. 西南交通大學物理科學與技術學院, 成都 610031; 2. 中國工程物理研究院核物理與化學研究所, 綿陽 621900;3.西南交通大學超導與新能源研究開發中心, 成都 610031)
采用基于第一性原理的密度泛函理論(DFT)和局域密度近似(LDA)方法,優化計算得到碳納米管(CNT),硼原子取代碳原子及其吸附氖原子前后系統的幾何結構,能量,電子能帶和態密度.結果顯示,碳納米管的能帶結構與石墨的層狀幾何結構相似,能量的變化只在kz=0和kz=0.5平面之間沿著c軸方向出現.B原子取代C原子使價帶和導帶分別分裂為兩個和三個能帶.對Ne原子的吸附使價帶能量沿著c軸方向升高并導致Fermi面附近的態密度下降.Ne原子的吸附在谷位H最穩定,頂位A其次.C-C間σ鍵的彎曲使Ne原子吸附在橋位b1比橋位b2處更為穩定.Ne原子在管外的吸附均為放熱過程,而管內則為吸熱過程.結構分析表明Ne原子對C原子有排斥作用,對B原子卻具有吸引作用.B原子取代C原子的位置略凸出于CNT的管壁之外,使Ne原子的吸附能增加.
碳納米管; 硼摻雜; Ne原子吸附
1 引 言
碳納米管(CNT)具有很好的物理特性,如高強度、低密度、高比表面、耐腐蝕性、導電性等[1-2].在碳納米管的表面或者內部可以生長金屬材料,獲得納米電子器件、磁性材料等[3].碳納米管在氣體的儲存、選擇性吸附等方面,都具有很好的特性和潛在的應用價值[4-5],因此在很多領域都得到廣泛的研究和利用.CNT可認為由單層石墨卷曲而成,具有很好的對稱性,且管內外電荷分布不相同.碳納米管對原子和分子具有很好的吸附性,如金屬和金屬離子[6-7],氧氣、氮氣[8-11]等.Seifollah[12]等人利用分子動力學的方法研究了Xe和Kr在開口碳納米管上的吸附,以及Xe在缺陷納米管上的吸附.Calbi[13]證明Xe、Ne和甲烷在碳納米管上的吸附.Tofighy[14]等研究了碳納米管對重金屬離子的吸附.Ackerman[15]研究Ar和Ne在納米管上的擴散率,比在硅質巖和沸石上高1至3個數量級. Majidi[16]研究了在22.67~49.82K下Ne在碳納米管上吸附.王昆鵬[17]等研究了B(N)摻雜對Al吸附的第一性原理研究,特別是B的摻雜具有良好的效果.Gong[18]等研究了碳納米管N摻雜的電催化作用.Zhou[19]研究了B(N)對儲氫性能的影響等.
氖氣在制造低溫環境、電光源、激光技術、高能物理等方面都有很廣泛的應用,但由于是惰性氣體,最外層電子數為8,結構穩定不易提取.目前多采用分離空氣再經液氫冷凝的方法獲得,但對溫度以及設備的要求比較高.碳納米管對稀有氣體的吸附已有理論和實驗驗證,但B摻雜對稀有氣體吸附是否有影響還未有報道.本文采用基于第一性原理的密度泛函理論(DFT)計算研究B摻雜CNT對Ne原子吸附的結構、能量、電子能帶和態密度的影響.
2 計算方法
使用基于密度泛函理論的晶體結構計算程序abinit[20],采用密度泛函理論(DFT)和電子交換關聯勢選用局域密度近似(LDA)方法,對CNT以及B摻雜的CNT-B系統吸附Ne原子前后的幾何結構和能量進行優化計算.計算中采用Troullier-Martins型的原子模守恒贗勢(NCPP)方法,C、B和Ne原子的電子組態為 2s22p2、2s22p1、2s22p6.在Brillouin區采用1×1×6,對k點進行抽樣,平面波的截斷能為20 Hartree,為了保證幾何優化的順利,平面波的截斷引入平滑拖尾參數0.5 Hartree,自洽場能量和原子力場的收斂閾值分別為1×10-6Hartree和2×10-3Hartree/Bohr.
3 結果與討論
3.1 幾何結構和電子能帶
采用圖1所示的手扶椅構型碳納米管單管SWCNT(4,4)作為計算模型,其原胞包含16個C原子.優化得到晶胞參數a=b=17.4257 ?,c=2.4457 ?,晶角α=β=90°,γ=120°,碳納米管單管之間的相互作用只有范德華力.B原子取代圖1中的碳原子C1后的晶胞參數為a=b=17.4261 ?,c=2.4718 ?.a、b基本不受B原子取代的影響,c增大了1.1%.
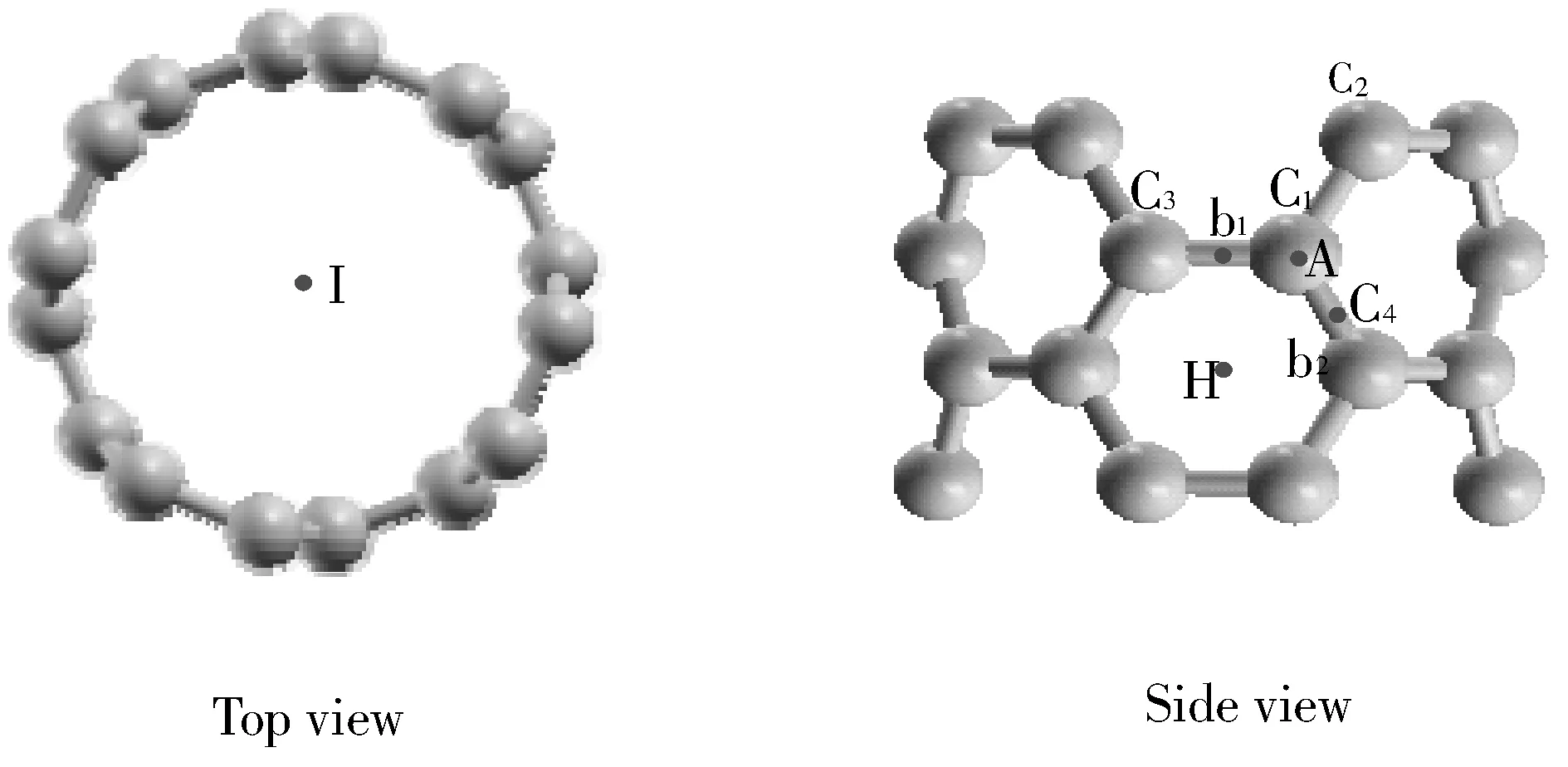
圖 1 手扶椅型碳納米管結構Fig.1 The structure of the armchair carbon nanotube
計算Ne原子的吸附時,采取圖1所示的1×1×2超胞結構.考慮到CNT結構的對稱性,Ne原子可能吸附在管外或者是管內.在管外的吸附位可能有,A位于C原子的頂位;H位于C六元環的中心谷位;b1位于與c軸垂直的C-C鍵的橋位;b2位于其它C-C鍵的橋位.Ne原子在管內的吸附位置為Ι.
圖2給出了CNT和CNT-B系統的能帶結構.有趣是,CNT具有相當于將石墨單層卷起來的幾何結構.原子的幾何空間坐標與反映動量的k坐標之間所具有的倒易關系,使CNT的能帶結構與石墨的層狀幾何結構相似.在布里淵區,kz=0和kz=0.5(相對坐標)的兩個平面,所有能帶的能量均分別保持不變.能帶在「-M-K以及H-A-L方向呈水平直線,能帶能量的變化都發生在沿著c軸的z方向.在Fermi面附近,能帶的變化顯示CNT沿著c軸方向具有導電特性.B原子取代C原子后形成的CNT-B系統與CNT比較,在「-M-K方向所代表的kz=0平面,價帶幾乎沒變化,而在Fermi面以上0.7eV處的空帶則分裂為了兩個能帶.在H-A-L方向所代表的kz=0.5平面,Fermi以下0.3eV處的價帶分裂為兩個能帶,而Fermi面以上1.2eV處的導帶則分裂為了三個能帶.在位于kz=0和kz=0.5平面之間的K-H和L-M方向,價帶與Fermi面均有所交叉,且在B原子取代C原子之后,價帶與上方的導帶之間出現約0.1eV的間隙.
圖2中的(b)和(d)顯示,Ne原子的吸附使CNT和CNT-B系統的Fermi面均有所下降.在Fermi面以下7.2eV處,Ne原子貢獻了一條平直的能帶.在kz=0的平面,被吸附的Ne原子使CNT系統的價帶與Fermi面非常接近,而Fermi面以上的導帶能量則從0.7eV顯著地升高到2.6eV.沿著c軸方向,價帶的能量顯著升高,與上方的導帶出現約0.2eV的能隙,價帶在kz=0平面的能量已達1.1eV.缺少電子的CNT-B系統吸附Ne原子,則進一步使價帶分裂出一條導帶,其能量在kz=0平面比Fermi面高0.5eV.價帶和導帶的能量沿著c軸方向同步升高,二者在kz=0.5平面的能量均比kz=0平面升高約1.0eV.
圖3為CNT和CNT-B系統的態密度.電子的極化使Ne原子具有供電性,CNT和CNT-B系統價帶的態密度均向左邊的低能方向移動.價帶和導帶的能量差值增加,Fermi面附近的態密度均出現顯著下降.態密度的這些變化使系統的導電性下降,但穩定性增強.
3.2 氖原子的吸附及其影響
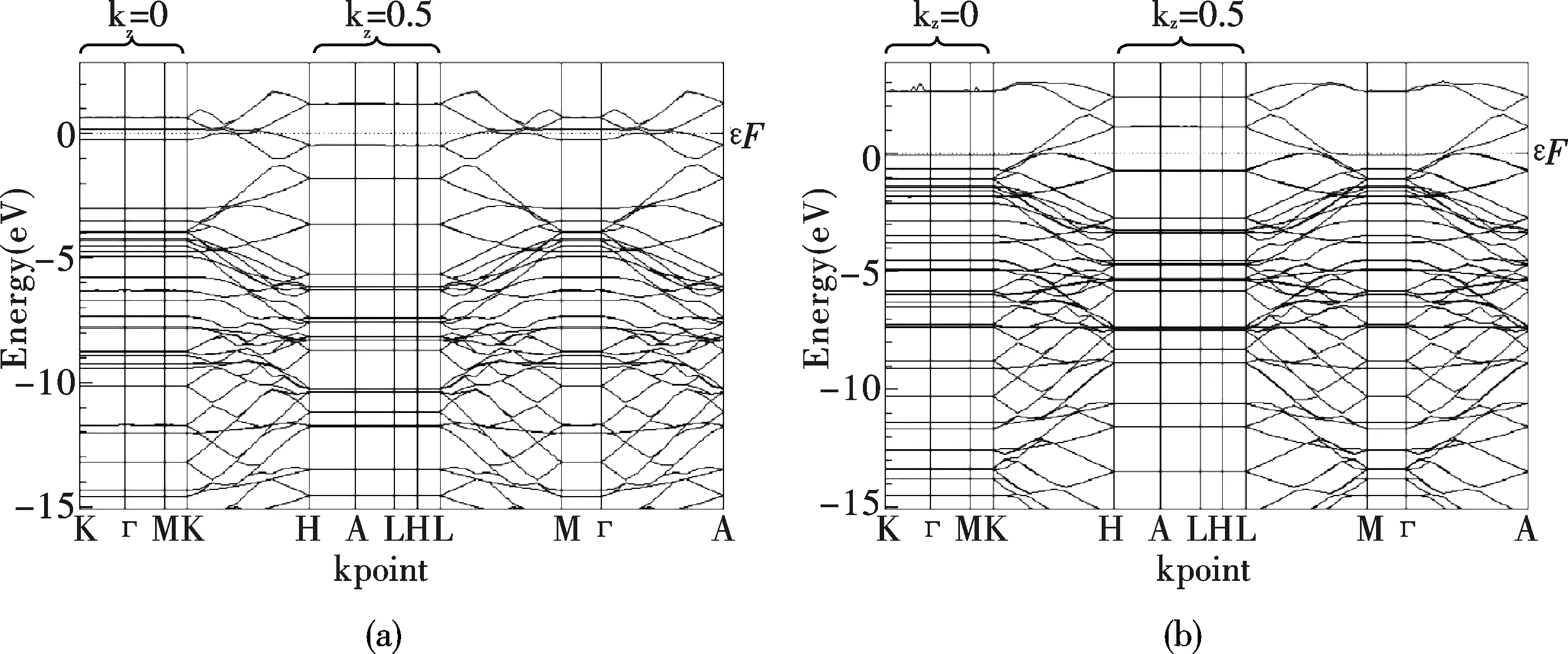
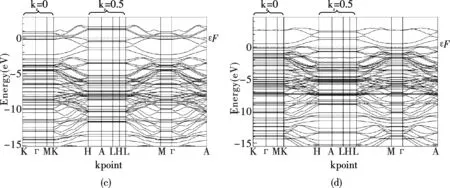
圖 2 電子能帶(a)CNT,(b)CNT吸附Ne原子,(c)CNT-B,(d)CNT-B吸附Ne原子Fig.2 Electronic energy bands of (a)CNT,(b)CNT adsorption neon atom, (c)CNT-B,(d)CNT-B adsorption neon atom
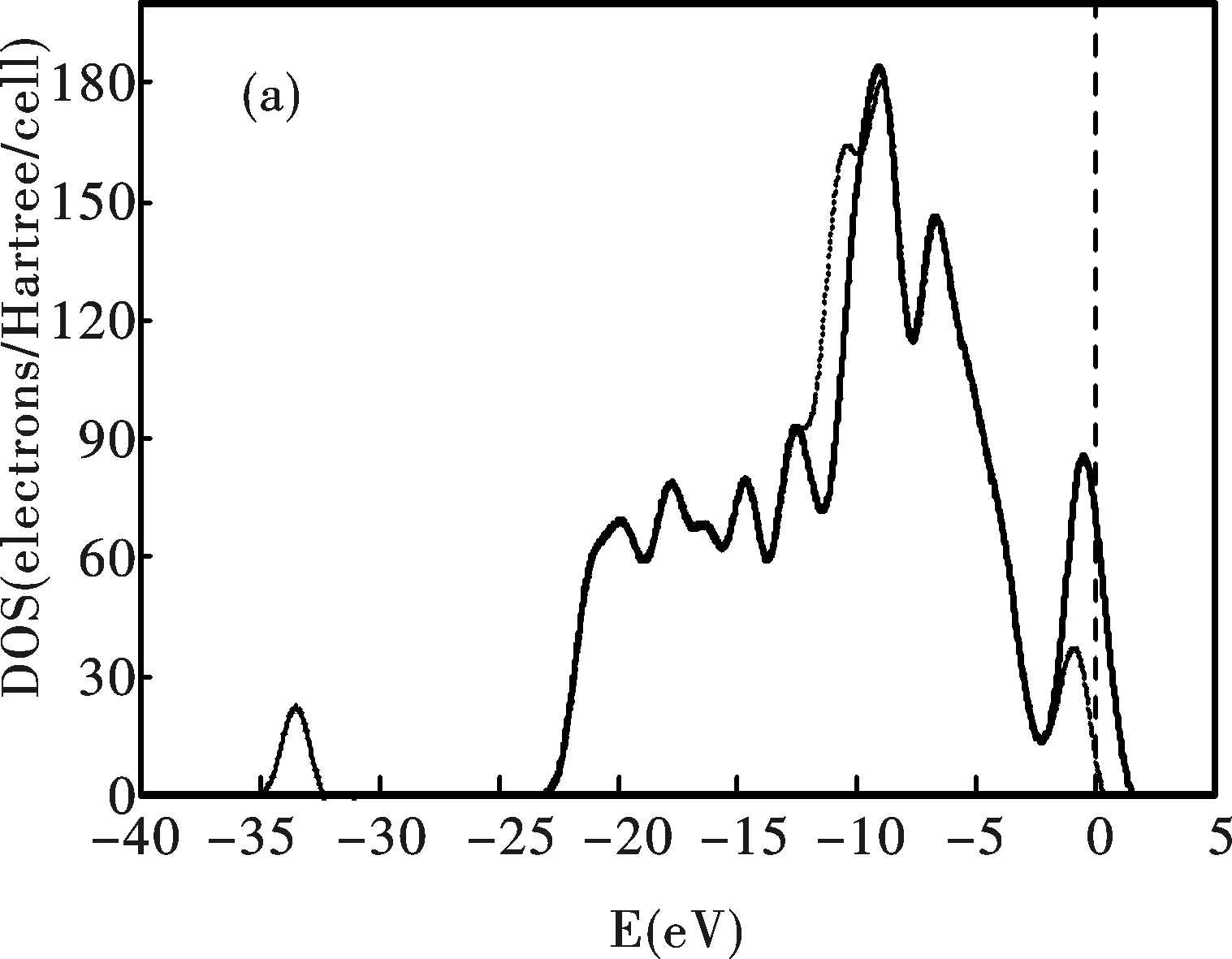
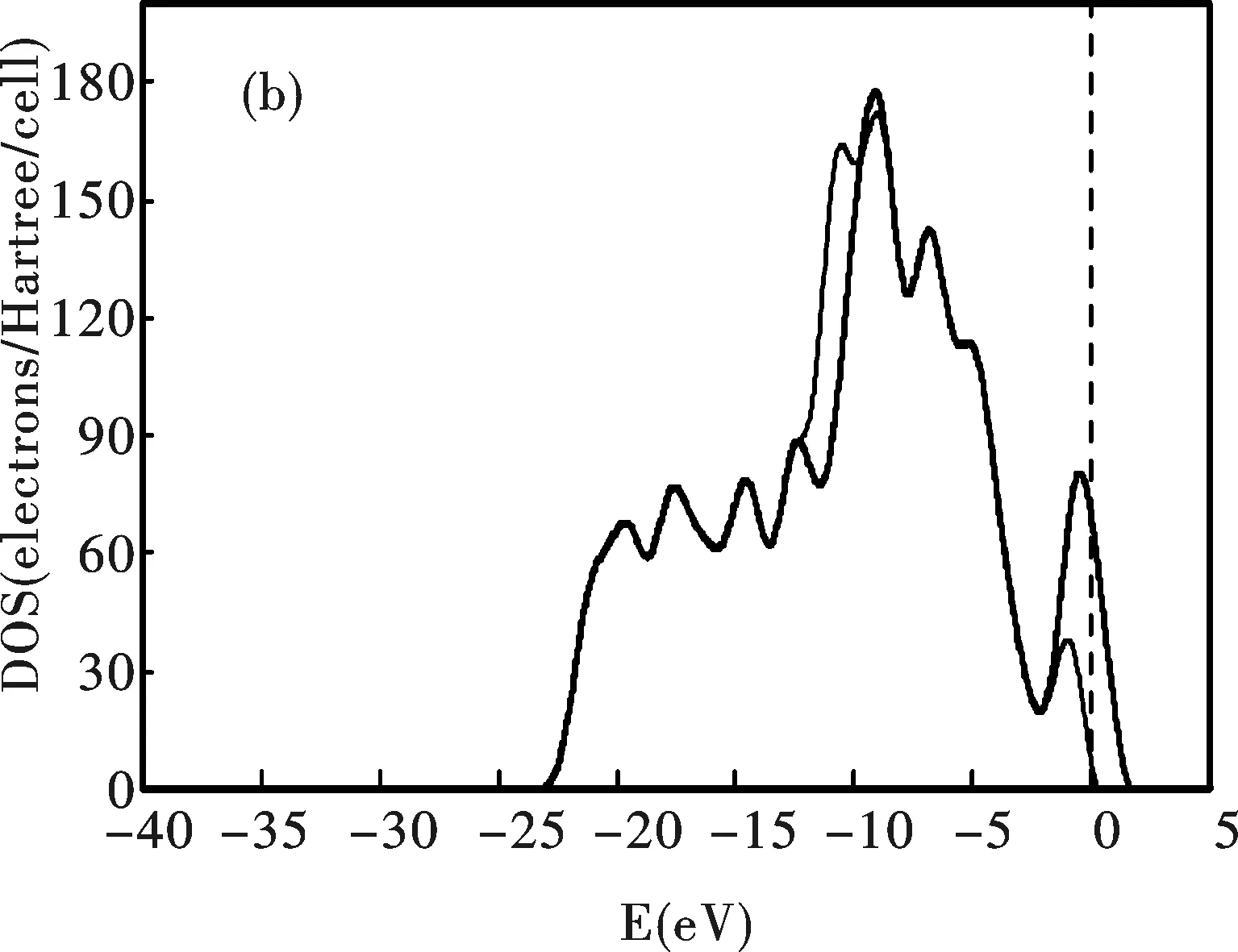
圖 3 CNT(a)和CNT-B(b)系統的電子態密度,虛線為吸附Ne以后的情形
表1給出了CNT吸附Ne前后的系統能量,以及B原子取代圖1中標注為C1的C原子對系統能量的影響.表2給出了吸附前后的吸附能,即Ea=ECNT-Ne-ENe-ECNT.Ea為負值時表明吸附為放熱過程,為正值則為吸熱過程.不難看出,Ne原子在CNT管外的吸附均為放熱過程,對吸附有利;在管內的吸附為吸熱過程,不利于吸附的實現.Ne原子吸附在管外電子密度相對較少的谷位H最穩定,吸附在C原子的頂位A穩定性其次,表明Ne原子的吸附更多地取決于具有較高對稱性的電場對核外電子極化的影響.兩個C原子之間的橋位b1和b2是C原子之間穩定的σ鍵中心,因而Ne原子在橋位b1和b2的吸附穩定性比谷位的H和頂位的A低.沿著CNT的周長方向,C1和C3之間的σ鍵彎曲性較大,其穩定性比C1和C2之間的σ鍵低,因此Ne原子吸附在橋位b1比橋位b2處更為穩定.在CNT內部,由于周圍的原子較多,電子密度較高,Ne原子受到較強的空間排斥.因此Ne原子在CNT內部I位的吸附能為正值.
B原子比C原子少一個價電子,當B取代圖1中C1位置的C原子時,周圍的電子密度下降.電子的極化有利于B周圍電子密度的增加,使B-C的σ鍵更穩定,Ne原子更容易被吸附.表2顯示B原子取代C原子之后,吸附Ne原子時的放熱均增加.
表3和表4分別給出了CNT和CNT-B系統吸附Ne原子前后的鍵長和鍵角.系統中C原子和B原子的價鍵軌道均為sp2雜化,成鍵軌道在CNT周長方向的彎曲,使鍵角小于sp2雜化的120°.周長方向C1-C3之間的σ鍵比其它方向的σ鍵略長,在CNT系統中二者相差0.4%.B原子取代C1處的C原子之后,σ鍵的長度均增加5%左右,且周長方向的σ鍵比其它方向的鍵長大1.3%.B原子周圍的鍵角比取代前減小了1.5%,表明B原子的位置略凸出于CNT的管壁之外.
吸附在CNT管壁外面的Ne原子對C原子有排斥作用.表3中可以看出, Ne原子吸附在頂位A時,C1位置的C原子被Ne原子排斥移向管內,與之相關的鍵角都有所增加.谷位H處Ne原子的排斥,使C1-C3鍵的彎曲度增加,鍵長縮短,系統沿著c軸方向延伸,∠C2C1C4增大,且斜向的C1-C2和C1-C4鍵增長.Ne原子在橋位b1時,σ鍵受Ne原子排斥的影響,彎曲度最大,對應的C1-C3的鍵長減少約0.3%;而Ne原子在橋位b2時,系統因Ne原子的排斥作用,沿著軸線方向伸長,∠C2C1C4增大,但鍵長幾乎不變.
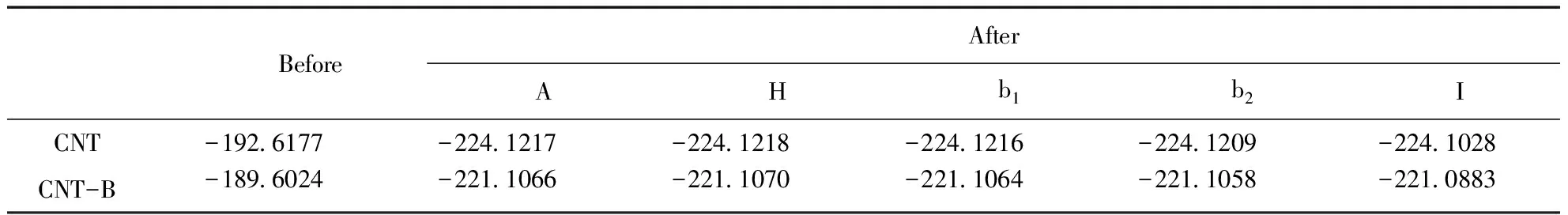
表1 CNT和CNT-B系統吸附Ne原子前后的體系能量(單位:Hartree)
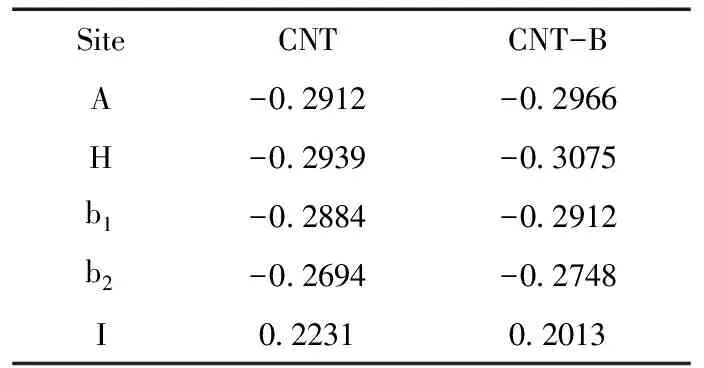
表 2 Ne原子在CNT和CNT-B系統壁外的吸附能(單位:eV)
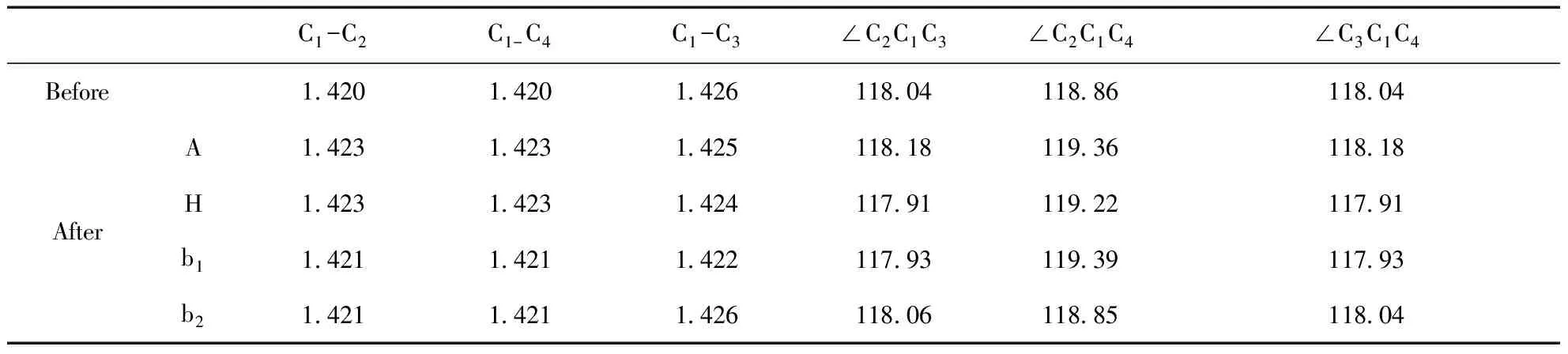
表 3 CNT系統吸附Ne原子前后的鍵長(單位:?)和鍵角(單位:度)
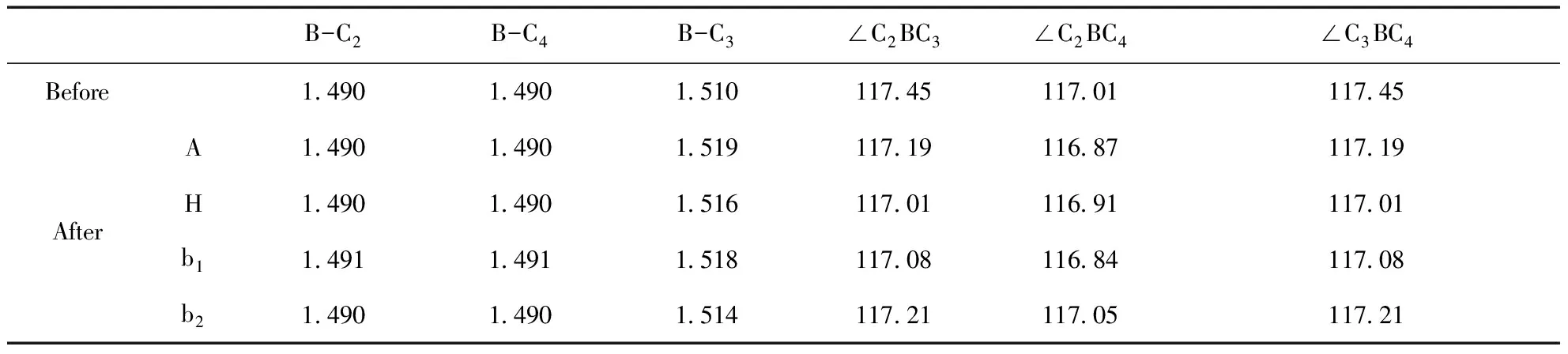
表 4 CNT-B系統吸附Ne原子前后的鍵長(單位:?)和鍵角(單位:度)
B原子本身凸出于CNT管壁之外,且價電子數比C原子少,對Ne原子具有吸引作用.B原子取代C1位置的C原子以后,其與周圍C原子的σ鍵長相應伸長.吸附在頂位A的Ne原子使B原子與C3位置的C原子的鍵長增加最為明顯,達到0.6%.Ne與B原子之間的相互吸引,使CNT-B系統在吸附Ne原子之后,鍵角均一般地呈減小之勢.
4 結 論
采用基于第一性原理的密度泛函理論(DFT)和局域密度近似(LDA)方法.優化計算得到碳納米管CNT,硼原子取代碳原子形成的CNT-B,及其吸附氖原子前后系統的幾何結構,能量,電子能帶和態密度.結果顯示,碳納米管的能帶結構與石墨的層狀幾何結構相似,在布里淵區,kz=0和kz=0.5(相對坐標)兩個平面方向,能帶的能量保持不變,呈水平直線,能量的變化發生在沿著c軸的z方向.B原子取代C原子對kz=0平面的價帶幾乎沒影響,kz=0.5平面的價帶和導帶則分別分裂為兩個和三個能帶.對Ne原子的吸附使價帶能量沿著c軸方向顯著升高,從而導致Fermi面附近的態密度顯著下降.
Ne原子在CNT管外的吸附均為放熱過程,而管內則為吸熱過程.Ne原子的吸附取決于電場對核外電子極化的影響,在管外的谷位H最穩定,頂位A其次.沿著CNT的周長方向,C-C間σ鍵的彎曲使Ne原子吸附在橋位b1比橋位b2處更為穩定.Ne原子在CNT內部I位的吸附能為正值.結果分析表明Ne原子對C原子有排斥作用,對B原子卻具有吸引作用.B原子取代C原子的位置略凸出于CNT的管壁之外,使Ne原子的吸附能增加.
[1] Xie S, Li W, Pan Z,etal. Mechanical and physical properties on carbon nanotube[J].JournalofPhysicsandChemistryofSolids, 2000, 61(7): 1153.
[2] Coleman J N, Khan U, Blau W J,etal. Small but strong: a review of the mechanical properties of carbon nanotube-polymer composites [J].Carbon, 2006, 44(9): 1624.
[3] Chen R J, Choi H C, Bangsaruntip S,etal.An investigation of the mechanisms of electronic sensing of protein adsorption on carbon nanotube devices [J].JournaloftheAmericanChemicalSociety, 2004, 126(5): 1563.
[4] Ye Y, Ahn C C, Witham C,etal. Hydrogen adsorption and cohesive energy of single-walled carbon nanotubes[J].AppliedPhysicsLetters, 1999, 74: 2307.
[5] Dillon A C, Jones K M, Bekkedahl T A,etal. Storage of hydrogen in single-walled carbon nanotubes[J].Nature, 1997, 386(6623): 377.
[6] Li Y H, Ding J, Luan Z,etal. Competitive adsorption of Pb2+, Cu2+and Cd2+ions from aqueous solutions by multiwalled carbon nanotubes[J].Carbon, 2003, 41(14): 2787.
[7] Che R, Peng L M, Duan X F,etal. Microwave absorption enhancement and complex permittivity and permeability of Fe encapsulated within carbon nanotubes[J].AdvancedMaterials, 2004, 16(5): 401.
[8] Sorescu D C, Jordan K D, Avouris P. Theoretical study of oxygen adsorption on graphite and the (8, 0) single-walled carbon nanotube[J].TheJournalofPhysicalChemistryB, 2001, 105(45): 11227.
[9] Bienfait M, Zeppenfeld P, Dupont-Pavlovsky N,etal.Thermodynamics and structure of hydrogen, methane, argon, oxygen, and carbon dioxide adsorbed on single-wall carbon nanotube bundles [J].PhysicalReviewB, 2004, 70(3): 035410.
[10] Yin Y F, Mays T, McEnaney B. Adsorption of nitrogen in carbon nanotube arrays [J].Langmuir, 1999, 15(25): 8714.[11] Babu D J, Lange M, Cherkashinin G,etal.Gas adsorption studies of CO2and N2in spatially aligned double-walled carbon nanotube arrays [J].Carbon, 2013, 61: 616.
[12] Simonyan V V, Johnson J K, Kuznetsova A,etal. Molecular simulation of xenon adsorption on single-walled carbon nanotubes[J].TheJournalofChemicalPhysics, 2001, 114: 4180.
[13] Calbi M M, Gatica S M, Bojan M J,etal. Phases of neon, xenon, and methane adsorbed on nanotube bundles [J].TheJournalofChemicalPhysics, 2001, 115: 9975.
[14] Tofighy M A, Mohammadi T. Adsorption of divalent heavy metal ions from water using carbon nanotube sheets [J].JournalofHazardousMaterials, 2011, 185(1): 140.
[15] Ackerman D M, Skoulidas A I, Sholl D S,etal. Diffusivities of Ar and Ne in carbon nanotubes[J].MolecularSimulation, 2003, 29(10-11): 677.
[16] Majidi R, Ghafoori Tabrizi K. Study of neon adsorption on carbon nanocones using molecular dynamics simulation [J].PhysicaB:CondensedMatter, 2010, 405(8): 2144.
[17] Wang K B, Shi C S, Zhao N Q,etal. First-principles calculations the properties that B (N)-doped single-walled carbon nanotubes adsorb aluminum atoms [J].Act.Phys.Sin., 2008, 57(12): 7833(in Chinese) [王昆鵬, 師春生, 趙乃勤, 等. B (N) 摻雜單壁碳納米管的Al原子吸附性能的第一性原理研究 [J].物理學報, 2008, 57(12): 7833]
[18] Gong K, Du F, Xia Z,etal. Nitrogen-doped carbon nanotube arrays with high electrocatalytic activity for oxygen reduction [J].Science, 2009, 323(5915): 760.
[19] Zhou Z, Gao X, Yan J,etal. Doping effects of B and N on hydrogen adsorption in single-walled carbon nanotubes through density functional calculations[J].Carbon, 2006, 44(5): 939.
[20] Gonze X, Beuken J M, Caracas R, Detraux F,etal. First-principles computation of material properties: the ABINIT software project [J].ComputationalMaterialsScience, 2002, 25: 478.
First-principle calculations for the adsorption of neon atom in carbon nanotube doped with boron atom
WEN Shu-Long1, WANG Qian2, PAN Min3, YAN Yue-Yang1, HUANG Zheng1
(1.School of Physical Science and Technology, Southwest Jiaotong University, Chengdu 610031, China;2. Institute of Nuclear Physics and Chemistry, China Academy of Engineering Physics, Mianyang 621900, China;3. Superconductivity and New Energy R&D Center Southwest Jiaotong University, Chengdu 610031, China)
Based on density functional theory (DFT) and local density approximation (LDA) methods, the geometries, the energies, electronic bands and density of states are optimized and calculated for the adsorption of neon atoms on carbon nanotubes (CNT) doped with boron atoms. The results show that the energy bands of CNT have a topology similar to the layered geometry of graphite. The energy changes of the bands between the two planes ofkz= 0 andkz= 0.5 occur only along the c-axis direction. The substituting boron atom splits the valence and conduction bands into two and three energy bands, respectively. The adsorption of neon atom makes the energy of the valence band rise along the c-axis direction, and the density of states near the Fermi surface decreases. The valley site H is the most stable site for the adsorption of neon atom, and the top site A is the second. The bending of the carbonic σ bond along the circumferential direction makes the bridge site b1more stable for the adsorption of neon than the bridge site b2. All the adsorptions of neon atoms outside of the CNT are exothermic; however that inside the tube endothermic. The analysis shows the repulsion of the neon on carbon atoms and the attraction to the boron atom. Protruding slightly beyond the wall of the CNT, the substituting boron atom makes the adsorption exothermic energy of neon atom increase.
Carbon nanotube; Adsorption of neon; Boron doping
103969/j.issn.1000-0364.2015.02.011
2013-11-06
中央高校基本科研業務費資助項目(swjtu12cx019)
溫述龍(1987—),男,河南濮陽人,碩士研究生,主要從事領域為稀有氣體的吸附.
黃整. E-mail: zhhuang@home.swjtu.edu.cn
O485
A
1000-0364(2015)02-0241-06
猜你喜歡
工業設計(2022年8期)2022-09-09 07:43:20
計算機應用(2022年2期)2022-03-01 12:33:42
計算機應用(2022年1期)2022-02-26 06:57:42
計算機應用(2021年4期)2021-04-20 14:06:36
計算機應用(2021年3期)2021-03-18 13:44:48
軍民兩用技術與產品(2021年10期)2021-03-16 06:05:30
計算機應用(2021年1期)2021-01-21 03:22:38
北京測繪(2020年12期)2020-12-29 01:33:58
裝備制造技術(2019年12期)2019-12-25 03:06:46
中國洗滌用品工業(2019年4期)2019-05-11 09:27:34
