不可逆性酪氨酸激酶抑制劑的研究進展
2015-04-21 06:41:32郭建軍趙永躍權騰飛苗震宇卜海之
中國藥理學通報 2015年6期
郭建軍,朱 晶,趙永躍,權騰飛,苗震宇,卜海之
(蘇州圣蘇新藥開發有限公司,江蘇蘇州 215104)
?
不可逆性酪氨酸激酶抑制劑的研究進展
郭建軍,朱晶,趙永躍,權騰飛,苗震宇,卜海之
(蘇州圣蘇新藥開發有限公司,江蘇蘇州215104)
摘要:酪氨酸激酶的過度表達和過度激活在許多腫瘤的發生和發展中具有重要意義,因此,多種酪氨酸激酶成為抗腫瘤藥物的靶點。目前已經上市的小分子酪氨酸激酶抑制劑多屬于可逆性抑制劑,這些藥物具有選擇性差、藥效不夠強烈和持久以及易引發耐藥性等缺點。近些年,不可逆性酪氨酸激酶抑制劑的研究正方興未艾。這一類藥物分子以不可逆的共價鍵與酪氨酸激酶上ATP結合域進行結合,從而使該靶點永久性失活。由于其獨特的作用機制,不可逆性酪氨酸激酶抑制劑可以有效地解決可逆性酪氨酸激酶抑制劑的幾個缺點。目前,已經有一批不可逆性酪氨酸激酶抑制劑進入市場或臨床研究階段。該篇綜述是對不可逆性酪氨酸激酶抑制劑的結構、藥理和藥化特征及其研究進展等進行總結和闡述。
關鍵詞:酪氨酸激酶;抑制劑;抗腫瘤藥物;不可逆;共價結合; ErbB; BTK
酪氨酸激酶在腫瘤的發生和發展中起到重要作用。科學家們已經開發出一系列的針對多種酪氨酸激酶的小分子化學藥物即“替尼”類藥物,并仍有相當數量的化合物處于臨床試驗階段。這些小分子酪氨酸激酶抑制劑(tyrosine kinase inhibitor,TKI)都以可逆性抑制的方式來發揮作用,由此帶來了一些缺點如選擇性不夠好、藥效不夠強烈和持久以及易引發耐藥性等。近些年,一類新的TKI-不可逆性TKI的研究正方興未艾。與可逆性TKI不同的是,這一類藥物作用的基礎是不可逆抑制[1]。不可逆性TKI的開發目前已被證實是解決可逆性TKI固有缺陷的有效途徑,現在進入到臨床試驗階段或最終上市的不可逆性TKI的數量正在快速增加。本篇綜述是對不可逆性酪氨酸激酶抑制劑的結構、藥理和藥化特征及其研究進展等進行總結和闡述。
1 可逆性和不可逆性TKI的一般藥理及藥化特征
目前,已上市的TKI中絕大部分都以可逆性結合于高度保守的ATP結合域(即占據“開關”部位)的方式,來阻止或減少酪氨酸激酶的磷酸化,最終實現抵抗腫瘤增殖的作用。這些可逆性抑制劑通常利用一個類似于ATP結構中嘌呤環(即ATP分子中與酪氨酸激酶結合的具體位置)的雜環如喹唑啉、喹啉、吲哚、吲唑等結構作為骨架核心來“模擬”ATP分子,以實現占據酪氨酸激酶中嘌呤環結合位點的目的。在該骨架核心上連接合適的疏水基團,可使分子與嘌呤環結合位點鄰近的疏水區域也發生作用,從而使該分子與整個ATP結合域達到更穩固、更具選擇性的結合。總體來講,這些可逆性抑制劑與ATP結合域的結合是通過較弱的、可逆性的作用力,如氫鍵、范德華力和疏水作用力等來實現的。這種較弱和可逆性的作用力基礎為這一類藥物帶來了3個不可忽視的缺點:①選擇性不夠好,②藥效不夠強烈和持久,③易引發耐藥性。所有蛋白激酶的ATP結合域都具有相似的空間結構和特性,這使得這一類藥物分子往往很難分辨“敵我”而與靶標之外的其它蛋白激酶進行不同程度的廣泛結合,這也就是所謂的“雜泛性”引起的“脫靶”作用。作用力的可逆性意味著,藥物分子在靶點處的濃度與其作用的強弱是即時相關的;而藥物分子在體內經受機體的清除,在其濃度不斷下降的同時靶標又可以不斷恢復活性,這就使得藥物在體內無法維持持久的藥效。作用力的可逆性同樣意味著,藥物分子在體內與ATP一起競爭性結合于靶標,而這種競爭性的存在可以使機體通過基因突變演化出更加傾向于與ATP結合的靶標蛋白突變體。
而不可逆性TKI通常以可逆性TKI的骨架結構為原型,在合適的位置連接上一個親電的功能團,如α,β-不飽和醛/酮結構,這個親電的功能團可以與ATP結合域附近的半胱氨酸殘基(富電子的親核結構)發生親電反應形成共價鍵[2]。與可逆性TKI相比,不可逆性TKI具有諸多獨特的優勢。首先,不可逆性TKI以永久性滅活的方式來發揮作用,這種“清剿”酶活性的方式當然使得其作用更為強烈而持久,即使藥物分子從循環系統中被完全清除掉,其藥效也仍能維持。其次,因為其和ATP與激酶的結合并不存在競爭性,也使得激酶突變的可能性降低而減輕或規避了耐藥性的產生[3]。最后,不可逆性TKI的選擇性非常高,因其分子結構上的親電功能團可以選擇性地與半胱氨酸殘基上的巰基反應。基因組學的研究發現,人類基因組編碼518種蛋白激酶(包括90種酪氨酸激酶),其中211種在其ATP結合域附近存在半胱氨酸殘基,這些半胱氨酸殘基出現的位置根據其激酶類別的不同而不同,科學家們可以針對這些不同位置上的非保守半胱氨酸殘基來設計和篩選出具有較高選擇性的不可逆性抑制劑[1,4]。這些抑制劑主要分為3類:①針對EGFR和BTK鉸鏈區附近半胱氨酸殘基的抑制劑[5-6];②針對FGFR ATP結合域頂部甘氨酸富集環上半胱氨酸殘基的抑制劑[7];③位于VEGFR ATP結合域底部的半胱氨酸殘基的抑制劑[8]。
因為具有可逆性TKI的骨架結構,不可逆性TKI通常也會保留有可逆性TKI的基本特點,如一定的選擇性和可逆性結合至ATP結合域的能力。這些特性對于不可逆性TKI是不可或缺的,因為:①藥物分子需要靠這種能力的引導來比較準確地定位至目標靶位,以此為基礎,親電功能團的存在才能更好地增強選擇性和藥效;②從安全性的角度來講,如果不存在這種初步選擇性,親電功能團的存在會使藥物分子更多地與非靶標蛋白質進行共價結合,從而極大地增加不可預期的毒性反應的風險。有鑒于此,不可逆性酪氨酸激酶抑制劑的結構優化需要對其可逆性抑制和親電反應的能力進行較好的平衡。
2 不可逆性ErbB抑制劑
目前進入市場和(或)臨床以及正在開發的不可逆性TKI中,針對EGFR(即ErbB1)和HER-2(即ErbB2)的占絕大多數。EGFR和HER-2是同屬于ErbB家族的受體型酪氨酸激酶,該家族還包括另外兩種激酶即ErbB3和ErbB4。在人類的許多癌癥如肺癌、頭頸癌、結腸直腸癌、卵巢癌、乳腺癌和膀胱癌中都發現了EGFR和HER-2的過度表達,這兩種激酶的高表達與不良的預后具有很大的關聯[9]。第1代針對ErbB的抗腫瘤靶向藥包括單靶點(EGFR)的治療非小細胞肺癌(NSCLC)的吉非替尼和厄洛替尼,以及雙靶點(EGFR和HER-2)的治療乳腺癌的拉帕替尼[10]。這3種藥都屬于可逆性抑制劑,它們在實際應用中都遇到了大量因獲得性耐藥性的生成而根本無法產生療效的臨床案例[11]。這種耐藥性的主要原因是這些可逆性抑制劑進入機體后,在靶點部位與ATP競爭而刺激機體生成了一些與ATP的親和力更強的EGFR和(或) HER-2蛋白的突變體,如T790M是野生型EGFR第790個氨基酸的位置上(ATP結合域附近)由絲氨酸突變為甲硫氨酸而來。T790M的生成是ErbB可逆性抑制劑治療過程中產生獲得性耐藥性的最主要原因,研究報道其與超過一半的獲得性耐藥性的案例有關[12]。
不可逆性ErbB抑制劑可與ATP結合域附近特定位置上的半胱氨酸殘基(EGFR上的Cys797、HER2上的Cys805 和ErbB4上的Cys803)發生不可逆的共價結合反應,而較少受到Thr790突變的影響,因此可以較好地規避這種突變帶來的獲得性耐藥性問題[13]。而且,在ATP結構域或其附近相應位置上(EGFR上的797位、HER2上的805位和ErbB4上的803位),半胱氨酸殘基的存在在其它所有激酶中都是幾乎沒有的,因此其構成了ErbB家族的一個高度特異性的結構特征。
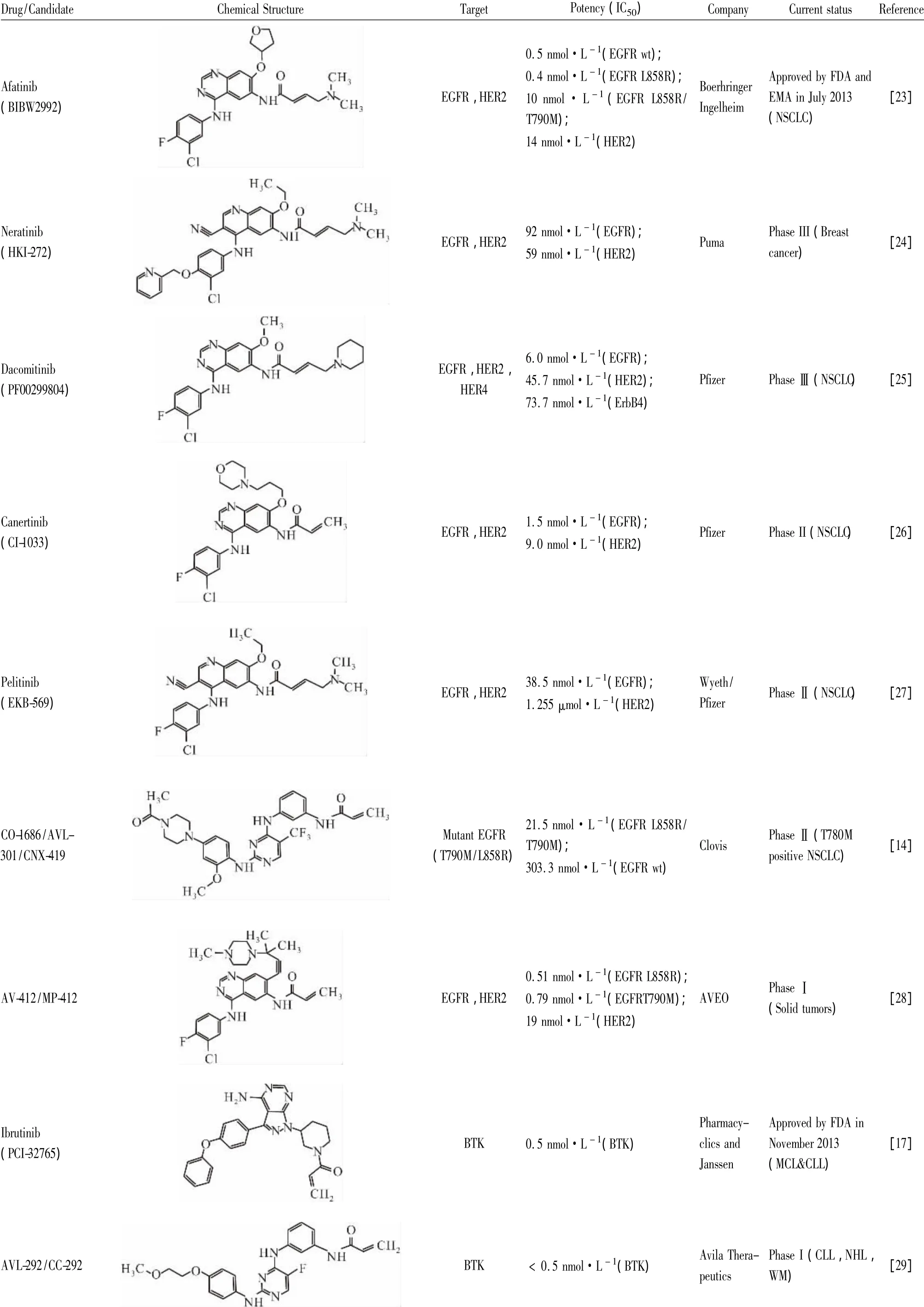
目前進入市場和(或)臨床試驗的不可逆性ErbB抑制劑總結如表1。大部分都以4-苯胺基喹唑啉(Fig 1,結構1) 和4-苯胺基-3-氰基喹啉(Fig 1,結構2)作為骨架的核心,引入親電的α,β-不飽和醛/酮結構,即邁克爾受體作為反應性功能團來與半胱氨酸上的巰基進行共價結合反應。前者有阿法替尼、達克替尼、卡奈替尼和AV-412,后者則有來那替尼和培利替尼。在這些抑制劑中,Boerhringer Ingelheim的阿法替尼已于2013年7月先后獲得FDA與EMA批準上市治療非小細胞肺癌(NSCLC),成為全球首個上市的不可逆性ErbB抑制劑,具有強烈的市場示范和提振作用。Puma公司于2014年向公眾宣布其從輝瑞公司收購的來那替尼在一項近期完成的臨床Ⅲ期試驗中獲得了成功。在Puma公司的報道中,來那替尼在治療HER2陽性乳腺癌的臨床研究中的表現優于羅氏的重磅藥物赫賽汀(NCT00878709)。這項臨床Ⅲ期研究的成功使得來那替尼上市的可能性大增。而輝瑞公司的達克替尼在兩項目前仍在進行的臨床III期的研究結果卻令人惋惜。據該公司披露,與對照藥厄洛替尼的比較中,達克替尼未能明顯改善NSCLC患者的疾病無進展生存期(NCT01360554) ;而在與與安慰劑的比較中,達克替尼未能明顯延長NSCLC患者的總生存期(NCT01000025)。輝瑞的卡奈替尼在2006年結束的一項臨床II期的研究中亦未能表現出對乳腺癌患者的臨床活性(NCT00051051),使得這一候選藥上市的前景黯淡。因此,卡奈替尼的后續臨床研究都已終止。由原惠氏公司(現被輝瑞公司并購)開發的培利替尼于2009年即結束了兩項分別針對結腸直腸癌(NCT00072748)和NSCLC(NCT00067548)的臨床Ⅱ期研究,但至今未見相關結果的正式報道,也未有后續臨床研究的開展。Clovis公司開發的CO-1686對突變EGFR具有較好的選擇性,其對T790M/L858R型和野生型EGFR的IC50分別為21.5和303.3 nmol·L-1[14]。目前,該抑制劑正在進行T790M突變型EGFR陽性的NSCLC患者群體上的兩項臨床Ⅱ期研究(NCT01526928和NCT02147990)。另外,AVEO公司開發的AV-412對突變型EGFR和HER2均具有很強的體外抑制作用,已于2011年完成一項在實體瘤患者身上進行的臨床I期研究(NCT00551850)。
另外,考慮到邁克爾受體的高度反應性所帶來的安全性隱患,科學家們也在嘗試利用其它反應性更低的親電基團或在生物體內通過轉化才能形成親電基團的結構來代替邁克爾受體[5]。前者如化合物3(Fig 1)的結構中含有炔基,并被證實可通過該炔基與EGFR共價結合[5]。后者如化合物4 (Fig 1),其本身并不具有親電性,但可在進入細胞后轉化為α,β-不飽和醛繼而發揮與EGFR共價結合的作用[16]。
3 不可逆性BTK抑制劑
BTK是非受體型酪氨酸激酶Tec家族的一員,幾乎完全由造血細胞表達。BTK在B細胞受體信號轉導通路(BCR)中起到至關重要的作用,同時也在前炎癥信號通路中起到介導作用。BTK抑制劑的開發也因此成為B淋巴細胞腫瘤和自身免疫性疾病治療的策略之一。與ErbB相似的是,BTK 在ATP結合域附近(即頂部甘氨酸富集環上)也存在一個半胱氨酸殘基Cys481[1,4],并且該半胱氨酸殘基也具有很高的選擇性。這一結構特征使得特異性的不可逆BTK抑制劑的發現與開發成為可能。
Zhengying等[16]在2007年的研究中就成功地發現了一系列活性很高的針對該處半胱氨酸殘基的BTK不可逆性抑制劑。這些化合物都是利用已經發現的對BTK有效的骨架結構,在其上連接了不同類型的邁克爾受體,其中的典型結構如Tab 1所示。Pharmacyclics公司通過一系列骨架結構的篩選發現,4’-氨基吡唑并[3,4-d]嘧啶化合物PCI-29732具有較強的BTK抑制作用(IC50=8.2 nmol·L-1),并在PCI-29732的基礎上引入α,β-不飽和醛形成一類不可逆性BTK抑制劑。通過體外藥效實驗發現這一類化合物,代表性的如依魯替尼(PCI-32765,IC50=0.5 nmol·L-1),都極大地增強了BTK的抑制作用[17]。依魯替尼已于2013年被FDA批準上市,成為BTK抑制劑的首創新藥,用于B細胞淋巴癌和慢性淋巴細胞白血病的治療。另一個針對Cys481的不可逆性BTK抑制劑,即由美國Celgene公司開發的AVL-292目前正處于臨床I期研究中,該化合物是在一個二氨基嘧啶的骨架上連接上一個α,β-不飽和醛[18],用于非霍奇金淋巴瘤和慢性淋巴細胞白血病的治療。
另外也有一些其它處于早期研究中的不可逆性BTK抑制劑被報導。如Locus Pharmaceuticals的科學家們發現了一個新的以吡咯并三嗪為特征的骨架結構可用來開發BTK抑制劑,并將α,β-不飽和醛引入至該結構形成化合物5(Fig 1),顯示出較強的BTK抑制(IC50=130 nmol·L-1)和B細胞分裂抑制作用[19]。輝瑞的科學家們則以咪唑并喹喔啉作為骨架核心,連接上α,β-不飽和醛而形成一系列的化合物,其中代表性的化合物6(Fig 1)具有很強的BTK抑制(IC50=1.93 nmol·L-1)作用[20]。
4 新生血管生成相關及其它的不可逆性酪氨酸激酶抑制劑
新生血管生成是指從預先存在的毛細血管以出芽方式形成新的血管的過程,這一過程是由多種因子來激發和調控的,包括VEGF、PDGF、bFGF等。其中,VEGF是目前發現的最為強大和專一的刺激血管內皮細胞增生的因子,而已發現 的VEGF受體(VEGFR)主要包括VEGFR-1(Flt-1)、VEGFR-2(Flk-1)和VEGFR-3(Flt-4)。以上三者均為受體型酪氨酸激酶,其中VEGFR-2主要分布在血管內皮細胞,是VEGF的主要功能受體,在新生血管生成的過程中起到關鍵性的作用。實體腫瘤的發生和發展都依靠大量的新生血管來輸送足夠營養成分,在胃癌、結腸癌、直腸癌、卵巢癌和乳腺癌等多數實體腫瘤組織中都發現了VEGFR表達水平的異常增高。
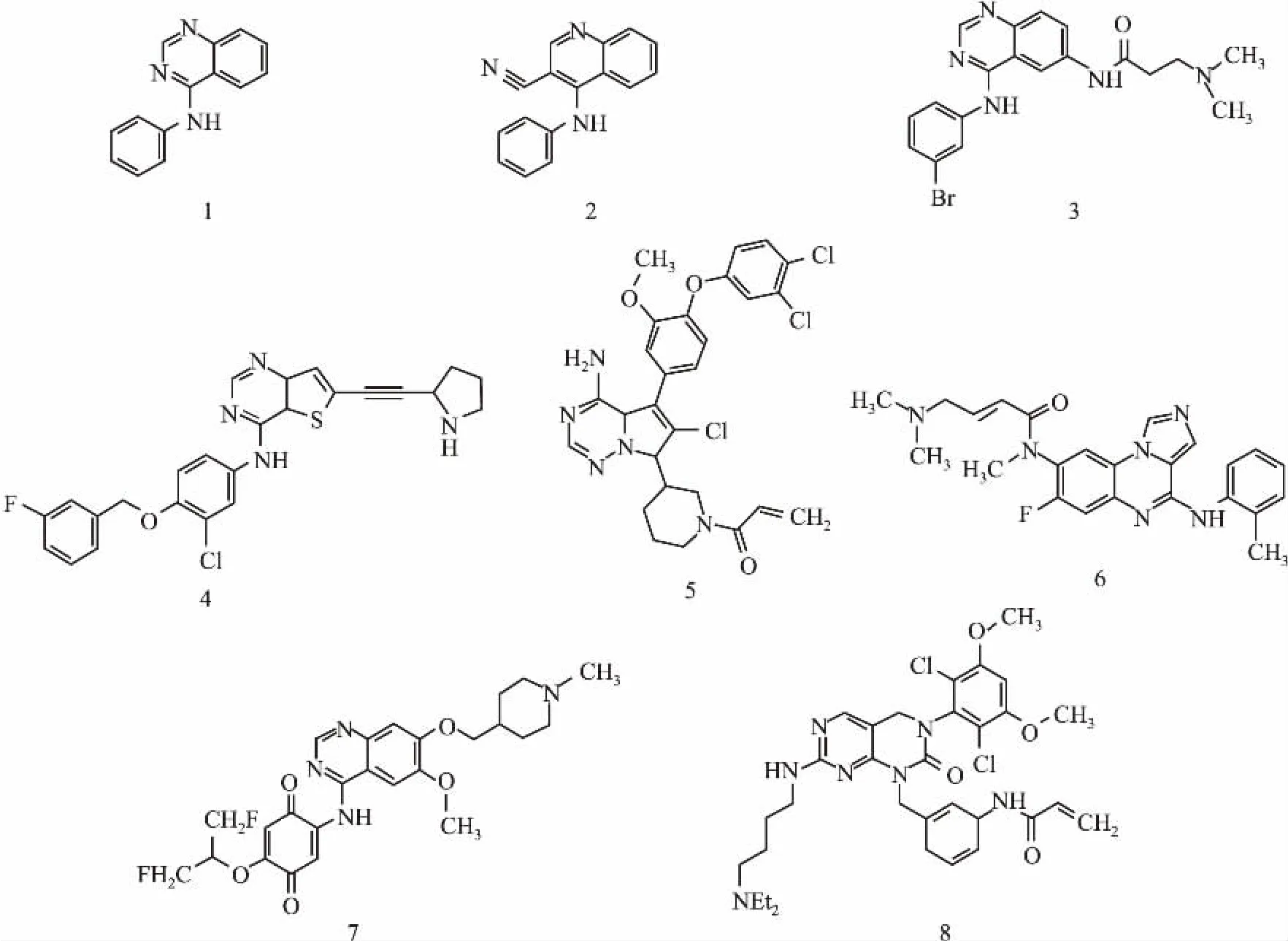
Fig 1 Structures of TKI drugs/candidates and relevant fragments
Tab 1 The information of TKI drugs or candidates in development
目前,市場上有多個針對VEGFR的可逆性抑制劑,如索拉非尼、舒尼替尼、帕唑帕尼、凡德他尼和阿西替尼等,但是這些藥物普遍存在選擇性不強的問題。這些VEGFR抑制劑同時也可對多種其它酪氨酸激酶產生強效抑制,如對PDGFR、c-kit、Flt3、RET、CSF1R、c-Raf和B-Raf等。低選擇性自然帶來了一些無可避免的副作用,科學家們也因此在不斷努力尋求新一代高選擇性的VEGFR抑制劑。所幸的是,與ErbB和BTK一樣,VEGFR也在ATP結合域附近(即結合域底部)存在一個特征性的半胱氨酸殘基Cys1045[8],這使得高選擇性不可逆性VEGFR抑制劑的開發成為選項之一。如Wissner等[21]在2005年尋找到一系列可與Cys1045形成共價結合的VEGFR-2抑制劑,如化合物7(Fig 1)。這些VEGFR抑制劑以喹唑啉為骨架的核心,在4-氨基上連接一個親電的醌結構,化合物7則是在醌上引入了具有強烈吸電子效應的氟,進一步增強了醌結構的親電性。體外細胞實驗表明該化合物可在納摩爾水平上對VEGFR-2產生抑制,并且在動物腫瘤模型上的研究也顯示出該化合物的抗腫瘤效果。
FGFR即bFGF的受體,屬于受體型酪氨酸激酶抑制劑,它是一個家族,包含4個成員,即FGFR-1、FGFR-2、FGFR-3 和FGFR-4。bGFR除了在新生血管生成中起到重要作用,也在腫瘤細胞增殖、遷移及分化等多重過程中起作用,也因此FGFR成為抗腫瘤的重要靶點之一。FGFR可逆性抑制劑如吉非替尼和厄洛替尼較差的體內藥理活性促使科學家們亦在進行不可逆性FGFR抑制劑的發現與開發。如Zhou等[22]在2010年即尋找到一個不可逆性FGFR抑制劑(化合物8,Fig 1),該化合物是在嘧啶并吡啶的核心骨架上連接了一個α,β-不飽和醛,實現了與Cys486的共價結合。體外藥效結果顯示,該化合物可抑制表達FGFR及其突變體的腫瘤細胞的分裂和生長[22]。
5 結語
不可逆性酪氨酸激酶抑制劑獨特的作用機制使之具有兩個非常突出的特征即選擇性強和藥理作用強烈而持久。因此,不可逆性酪氨酸激酶抑制劑可以減輕或避免因與ATP競爭而引起的激酶突變體的生成,從而減輕或避免獲得性耐藥性的產生。但與此同時,不可逆性酪氨酸激酶抑制劑分子中的反應性結構域的存在也帶來了更多的因“脫靶作用”而造成的毒副作用的擔憂。目前,已經有一批不可逆性酪氨酸激酶抑制劑進入市場或臨床研究階段,其中一些如阿法替尼、依魯替尼和來那替尼的臨床數據顯示這一類化合物在保留較好藥效作用的前提下亦能達到很好的安全性。這說明不可逆性酪氨酸激酶抑制劑是一個非常值得繼續研究和開發的領域。但是我們也需要認識到,這一類藥物的開發仍然處于初期階段,科學家們仍然需要付出更多的努力來完善其理論基礎,同時也需要嘗試更多不同的親電功能團和共價結合策略以及得到更多的臨床研究數據來更可靠地驗證這一類藥物的安全性。
參考文獻:
[1]Singh J,Petter R C,Baillie T A,Whitty A.The resurgence of covalent drugs[J].Nat Rev Drug Discov,2011,10(4) :307-17.
[2]Singh J,Petter R C,Kluge A F.Targeted covalent drugs of the kinase family[J].Curr Opin Chem Biol,2010,14(4) :475-80.
[3]Doebele R C,Oton A B,Peled N,et al.New strategies to overcome limitations of reversible EGFR tyrosine kinase inhibitor therapy in non-small cell lung cancer[J].Lung Cancer,2010,69(1) : 1-12.
[4]Leproult E,Barluenga S,Moras D,et al.Cysteine mapping in conformationally distinct kinase nucleotide binding sites: application to the design of selective covalent inhibitors[J].J Med Chem,2011,54(5) :1347-55.
[5]Carmi C,Lodola A,Rivara S,et al.Epidermal growth factor receptor irreversible inhibitors: chemical exploration of the cysteinetrap portion[J].Mini Rev Med Chem,2011,11(12) :1019-30.
[6]Lou Y,Owens T D,Kuglstatter A,et al.Bruton’s tyrosine kinase inhibitors: approaches to potent and selective inhibition,preclinical and clinical evaluation for inflammatory diseases and B cell malignancies[J].J Med Chem,2012,55(10) :4539-50.
[7]Zhou W,Hur W,McDermott U,et al.A structureguided approach to creating covalent FGFR inhibitors[J].Chem Biol,2010,17 (3) :285-95.
[8]Barluenga S,Jogireddy R,Koripelly G K,Winssinger N.In vivo efficacy of natural product-inspired irreversible kinase inhibitors [J].Chem Bio Chem,2010,11(12) :1692-9.
[9]Temam S,Kawaguchi H,El-Naggar A K,et al.Epidermal growth factor receptor copy number alterations correlate with poor clinical outcome in patients with head and neck squamous cancer[J].J Clin Oncol,2007,25(16) :2164-70.
[10]陶黎陽,符立梧.EGFR/HER-2雙受體酪氨酸激酶抑制劑拉帕替尼的研究進展[J].中國藥理學通報,2008,24(12) :1541 -4.
[10]Tao L Y,Fu L W.Progress of study on lapatinib[J].Chin Pharmacol Bull,2008,24(12) :1541-4.
[11]Rosenzweig SA.Acquired resistance to drugs targeting receptor tyrosine kinases[J].Biochem Pharmacol,2012,83(8) : 1041-8.
[12]劉姣,李明春.PI3K/Akt通路與表皮生長因子受體酪氨酸激酶抑制劑產生耐藥性的關系研究進展[J].中國藥理學通報,2013,29(12) :1648-50.
[12]Liu J,Li M C.PI3K/Akt signaling pathway and resistance to epithelial growth factor receptor tyrosine kinase inhibitors[J].Chin Pharmacol Bull,2013,29(12) :1648-50.
[13]Blair J A,Rauh D,Kung C,et al.Structure-guided development of affinity probes for tyrosine kinases using chemical genetics[J].Nat Chem Biol,2007,3(4) :229-38.
[14]Walter A O,Sjin R T,Haringsma H J,et al.Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M-mediated resistance in NSCLC[J].Cancer Discov,2013,3(12) : 1404-15.
[15]Carmi C,Galvani E,Vacondio F,et al.Irreversible inhibition of epidermal growth factor receptor activity by 3-aminopropanamides [J].J Med Chem,2012,55(5) :2251-64.
[16]Pan Z,Scheerens H,Li S J,et al.Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase[J].Chem Med Chem,2007,2(1) :58-61.
[17]Honigberg L A,Smith A M,Sirisawad M,et al.The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy [J].Proc Natl Acad Sci USA,2010,107(29) :13075-80.
[18]Kluge A,Petter R C,Tester R W,et al.Heteroaryl compounds and uses thereof[P].PCT Int Appl WO,2009158571;2009.
[19]Konteatis Z,Moffett K,Lee Y,Chao W.Pyrrolotriazine compounds[P].Patent Application WO 2010/126960,2010.
[20]Pan Z,Li S J,Schereens H,et al.Inhibitor-derived reporter molecules for detection of bruton’s tyrosine kinase and their uses[P].Patent Application WO 2008/054827,2008.
[21]Wissner A,Floyd M B,Johnson B D,et al.2-(Quinazolin-4-ylamino) -[1,4]benzoquinones as covalent-binding,irreversible inhibitors of the kinase domain of vascular endothelial growth factor receptor-2[J].J Med Chem,2005,48(24) :7560-81.
[22]Zhou W,Hur W,McDermott U,et al.A structure guided approach to creating covalent FGFR inhibitors[J].Chem Biol,2010,17(3) :285-95.
[23]Li D,Ambrogio L,Shimamura T,et al.BIBW2992,an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models[J].Oncogene,2008,27(34) :4702-11.
[24]Rabindran S K,Discafani C M,Rosfjord E C,et al.Antitumor activity of HKI-272,an orally active,irreversible inhibitor of the HER-2 tyrosine kinase[J].Cancer Res,2004,64(11) : 3958-65.
[25]Engelman J A,Zejnullahu K,Gale C M,et al.PF00299804,an irreversible pan-ERBB inhibitor,is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib [J].Cancer Res,2007,67(24) :11924-32.
[26]Smaill J B,Rewcastle G W.Tyrosine kinase inhibitors.17.Irreversible inhibitors of the epidermal growth factor receptor: 4-(phenylamino) quinazoline-and 4-(phenylamino) pyrido[3,2-d]pyrimidine-6-acrylamides bearing additional solubilizing functions[J].J Med Chem,2000,43(7) :1380-97.
[27]Torrance C J,Jackson P E,Montgomery E,et al.Combinatorial chemoprevention of intestinal neoplasia[J].Nat Med,2000,6 (9) :1024-8.
[28]Suzuki T,Fujii A,Ohya J,et al.Pharmacological characterization of MP-412 (AV-412),a dual epidermal growth factor receptor and ErbB2 tyrosine kinase inhibitor[J].Cancer Sci,2007,98(12) : 1977-84.
[29]Evans E,Tester R,Aslanian S.Clinical development of AVL-292: A potent,selective covalent btk inhibitor for the treatment of B cell malignancies[C].ASH Annual Meeting December 10-13,2011 San Diego,CA.
Research progress on irreversible tyrosine kinase inhibitors
GUO Jian-jun,ZHU Jing,ZHAO Yong-yue,QUAN Teng-fei,MIAO Zhen-yu,BU Hai-zhi
(3D BioOptima Co.,Ltd.,Suzhou Jiangsu 215104,China)
Abstract:Dysfunction in tyrosine kinase activity disrupts the normal control of cellular phosphorylation signaling pathways,which plays a vital role in genesis and development of various tumors,and makes tyrosine kinases a class of targets of many anti-tumor drugs.Currently most approved tyrosine kinase inhibitors (TKIs) are based on irreversible binding mechanisms,making them poorly selective,not potent or sustained enough regarding pharmacological effects and prone to triggering resistance.In the past decade,much progress has been made in the development of a new class of TKIs which irreversibly inhibit their target proteins via the formation of covalent bonds,overcoming the drawbacks of irreversible TKIs.Several irreversible TKIs have entered markets or clinical research phases.This review is to summarize the structural,pharmacological and medicinal chemical properties of investigational and marketed irreversible TKIs as well as their recent developments.
Key words:tyrosine kinase; inhibitor; anti-tumor drug; irreversible; covalent binding; ErbB; BTK
作者簡介:郭建軍(1980-),男,博士,研究方向:藥理學,Tel: 0512-67683273,E-mail: jianjun.guo@3dbiooptima.com;卜海之(1962-),男,博士,研究方向:藥物代謝動力學,通訊作者,Tel: 0512-67683273,E-mail: haizhi.bu @ 3dbiooptima.com
基金項目:蘇州科學技術局姑蘇創新創業領軍人才基金的資助(No XG0826)
收稿日期:2015-02-12,修回日期:2015-03-20
文獻標志碼:A
文章編號:1001-1978(2015) 06-0749-06中國圖書分類號: R-05; R345.57; R979.1; R977.3
doi:10.3969/j.issn.1001-1978.2015.06.003
