高效液相色譜法測定愈傷靈膠囊中黃曲霉毒素G2,G1,B2,B1含量*
2015-05-31 02:51:46張正鋒
中國藥業 2015年15期
張正鋒,張 毅
(重慶市食品藥品檢驗所·重慶市藥物過程與質量控制工程技術研究中心,重慶 401121)
中藥材及其制劑在加工、運輸、儲存過程中經常會發生霉變現象,產生霉菌毒素,其中以黃曲霉毒素的毒性最大。黃曲霉毒素已被世界衛生組織的癌癥研究中心確定為一類致癌物,其毒性為氰化鉀的10倍,砒霜的68倍。當前世界各國已在很多食品、藥品標準中都制訂了嚴格的黃曲霉毒素殘留限度,2010年版《中國藥典(一部)》已開始對中藥材中黃曲霉毒素進行控制,并收載了黃曲霉毒素G2,G1,B2,B1的檢測方法。愈傷靈膠囊為傳統中成藥,處方包括土鱉蟲、續斷、三七、當歸、自然銅等藥味,其中土鱉蟲可能污染黃曲霉毒素[1],且土鱉蟲、當歸、三七都是原生藥粉入藥,因此有必要制訂黃曲霉毒素檢查項,以控制產品質量,保證用藥安全。《中國藥典(一部)》只對桃仁、胖大海、陳皮、僵蠶等中藥材規定了限度,香港衛生署公布的中藥材標準中對當歸、三七制訂了黃曲霉毒素的限量檢查[2]。近年來越來越多的研究已經發現,中成藥中也有黃曲霉毒素殘留[3-5]。本研究中按照2010年版《中國藥典(第二增補本)》附錄中黃曲霉毒素殘留量測定法[6],采用免疫親和柱凈化-高效液相色譜(HPLC)-柱后光化學衍生-熒光檢測技術,測定愈傷靈膠囊中黃曲霉毒素G2,G1,B2,B1的殘留量,并對陽性樣品采用超高效液相色譜-串聯質譜(UPLC-MS/MS)法進行了驗證。
1 儀器與試藥
Waters2695型高效液相色譜儀(Waters2475熒光檢測器<美國Waters公司>,光化學衍生器<美國VICAM公司,紫外光燈254 nm>,Waters ACQUITY TM串連四級桿質譜儀<MassLynx工作站>);免疫親和層析柱(北京華安麥科生物技術有限公司);MSU224S-100-DU型電子天平(德國Sartorius公司);超聲波清洗器(昆山市超聲波儀器有限公司);Z2064型離心機(德國HERMLE公司);Milli-Q超純水處理系統。愈傷靈膠囊為售樣品;甲醇、乙腈均為色譜純,水為高純水;黃曲霉毒素混合標準品(黃曲霉毒素B1,B2,G1,G2質量濃度分別為 1.04,0.35,1.18,0.59 μg/mL,中國食品藥品檢定研究院,批號為610001-201301)。
2 方法與結果
2.1 色譜條件
2.1.1 HPLC
色譜柱:DikmaDiamonsilC18柱(250mm ×4.6 mm,5 μm);柱溫:40℃;流動相:甲醇(A)-乙腈(B)-水(C),梯度洗脫(0~17 min,A 25% →45% ,B 10% ,C 65% →45%;17~18 min,A 45% →65%,B 10%,C 45%→25%;18 ~23 min,A 65%,B 10%,C 25%;23~24 min,A 65% →25% ,B 10% ,C 25% →65%);流速:1.1 mL/min;進樣量:30 μL。采用柱后光化學衍生法:光化學衍生器(254 nm);以熒光檢測器檢測,激發波長λex=360 nm,發射波長λem=450 nm。色譜見圖1。
2.1.2 UPLC - MS/MS
色譜柱:Waters ACQUITY UPLC BEH C18柱(50 mm ×2.1mm,1.7 μm);柱溫:30 ℃;流速:0.4 mL/min;進樣量:5 μL;流動相:A為甲醇-乙腈(3∶2),B為0.1%乙酸水溶液,梯度洗脫(0~5 min,A 40% →70% ,B 60% →30%;5~6 min,A 70% ,B 30%;6 ~6.5 min,A 70% →40% ,B 30% →60%);ESI正離子 MRM 模式:離子源溫度120℃,毛細管電壓3.0 kV,脫溶劑氣溫度350℃,脫溶劑氣流速500 L/h;鞘氣流速50 L/h;定性離子對及相關參數見表1。
2.2 溶液制備

圖1 高效液相色譜圖
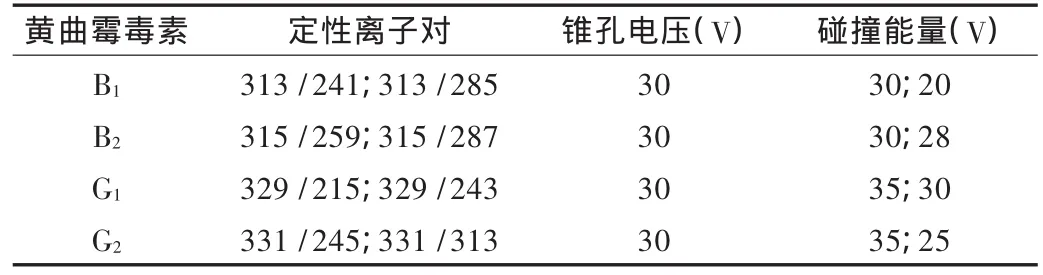
表1 UPLC-MS/MS法定性離子對、錐孔電壓及碰撞能量
精密量取黃曲霉毒素混合標準品0.5 mL,置10 mL容量瓶中,用甲醇稀釋至刻度,作為貯備液(黃曲霉毒素B1質量濃度為50 ng/mL)。精密量取貯備液1 mL,置25 mL容量瓶中,用甲醇稀釋至刻度,即得混合對照品溶液(黃曲霉毒素B1質量濃度為2 ng/mL)。取供試品粉末5 g,精密稱定,加氯化鈉3 g,精密加入70%甲醇溶液50 mL,振搖使溶散,超聲處理30 min,離心5 min(離心速度4 000 r/min),精密量取上清液10 mL,置50 mL容量瓶中,加水約 25 mL,振搖,放冷,用水稀釋至刻度,搖勻,用 0.45 μm微孔濾膜濾過;精密量取20 mL,通過免疫親合層析柱,流速為3 mL/min,再用水10 mL洗滌,洗液棄去,使空氣進入柱子,將水擠出柱子,以1.5 mL甲醇以重力作用自然洗脫,收集洗脫液至2 mL量瓶中,并用甲醇稀釋刻度,搖勻,即得供試品溶液。取70%甲醇溶液50 mL,按供試品溶液制備方法下操作,即得空白溶液。
2.3 方法學考察
線性關系考察:取黃曲霉毒素混合對照品溶液,分別進樣3,5,10,15,20,25,30 μL,以各色譜峰峰面積(Y)對黃曲霉毒素絕對量(X)進行回歸,得回歸方程。結果見表2。

表2 黃曲霉毒素線性范圍
精密度試驗:將混合對照品溶液(黃曲霉毒素B1質量濃度為2 ng/mL)連續進樣 6 次,依法測定。結果黃曲霉毒素 G2,G1,B2,B1的 RSD 分別為 1.29% ,1.45% ,1.88% ,1.51%(n=6),表明儀器精密度良好。
穩定性試驗:取不含黃曲霉毒素的樣品粉末5 g,精密稱定,精密加入黃曲霉毒素混合對照品貯備液0.1 mL,依法制備供試品溶液,分別于 0,4,8,12 h進樣,依法測定。結果黃曲霉毒素 G2,G1,B2,B1含量的 RSD 分別為 1.43% ,1.09% ,1.58% ,1.09%(n=4),表明供試品溶液在12 h內穩定。
重復性試驗:取不含黃曲霉毒素的樣品粉末5 g,共6份,精密稱定,精密加入黃曲霉毒素混合對照品貯備液0.1 mL,依法制備供試品溶液并測定。結果黃曲霉素G2,G1,B2,B1的 RSD分別為8.0% ,10.9% ,8.9%,6.2%(n=6),表明該法重復性良好[2]。
加樣回收試驗:對該方法進行三水平加樣回收試驗。添加濃度以黃曲霉毒素B1計分別為1,5,10 μg/kg。取不含黃曲霉毒素的樣品粉末5 g,精密稱定,共取9份,分別精密加入混合對照品貯備液 0.1,0.5,1.0 mL,依法制備供試品溶液,測定含量,計算回收率。結果見表3。參照相關技術要求,回收率范圍應為50% ~120%,RSD<15%[2],表明該法的加樣回收試驗結果符合要求。
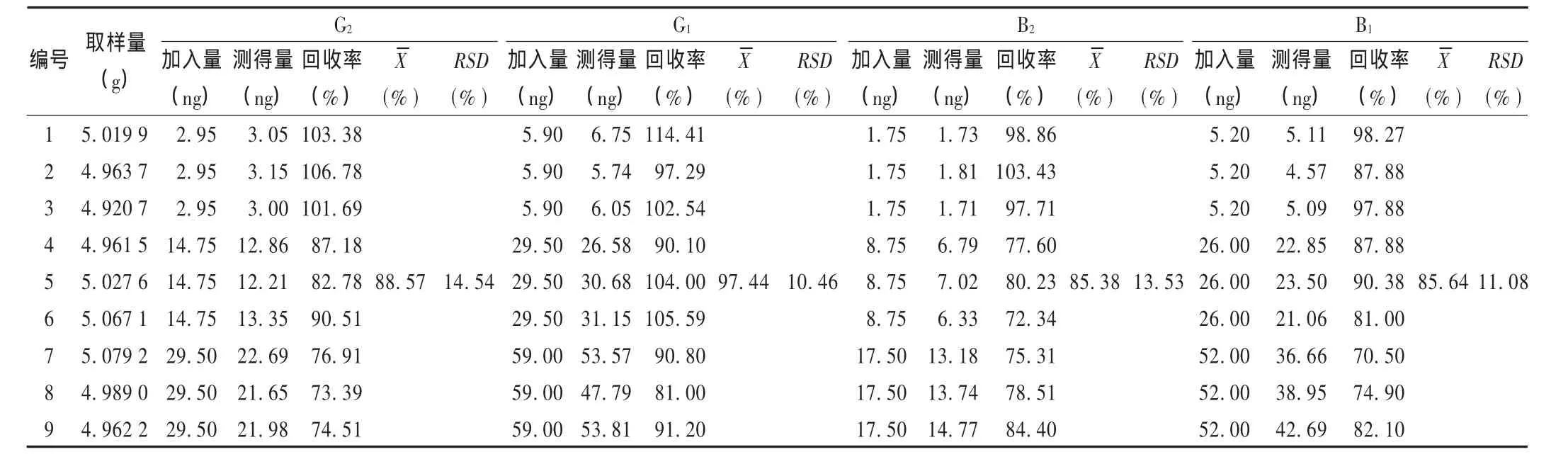
表3 黃曲霉毒素G2,G1,B2,B1加樣回收試驗結果(%,n=9)
檢出限與定量限確定:將上述加樣回收試驗的供試品溶液稀釋成適當濃度進樣分析,檢出限為信噪比(S/N)=3,定量限為S/N=10,則以黃曲霉毒素B1計,儀器檢出限為0.25 pg,定量限為 0.83 pg,本法的定量限為 0.14 μg/kg。黃曲霉毒素 B2,G1,G2按此方法得到的定量限分別為 0.06,0.21,0.13 μg/kg。
2.4 樣品含量測定
結合線性、加樣回收率及儀器檢出限與定量限測定,本試驗報告限暫訂為:黃曲霉毒素 B1,B2,G1,G2及其總量(B1+B2+G1+G2)均為0.5 μg/kg,低于報告限的視為未檢出。測定了82批愈傷靈膠囊樣品,結果49批樣品檢出黃曲霉毒素,黃曲霉毒素總量≥10 μg/kg的有 9 批,其中 6 批黃曲霉毒素 B1≥5 μg/kg,見圖 2(僅列出陽性樣品黃曲霉毒素B1及總量)。
2.5 陽性樣品確證試驗

圖2 陽性樣品含量測定結果
黃曲霉毒素B1,G1在含水的溶劑中發生熒光淬滅,經波長254 nm的紫外光燈照射后,形成具有穩定熒光的物質,即是柱后光化學衍生的原理。故采用衍生與未衍生時黃曲霉毒素的峰面積比較,即可簡單地對陽性樣品進行初步確認。本試驗結果顯示,衍生后黃曲霉毒素B1的峰面積約為未衍生時的9倍,G1的峰面積約為未衍生時的12倍,與郝愛魚等[7]報道基本一致。以每1成分選取2對靈敏度最高的子母離子,采用MRM模式檢測,樣品在與對照品相同保留時間處同時檢出2對離子對,且其強度的比值與對照品一致,即可確證樣品中含有黃曲霉毒素。MRM圖見圖3。
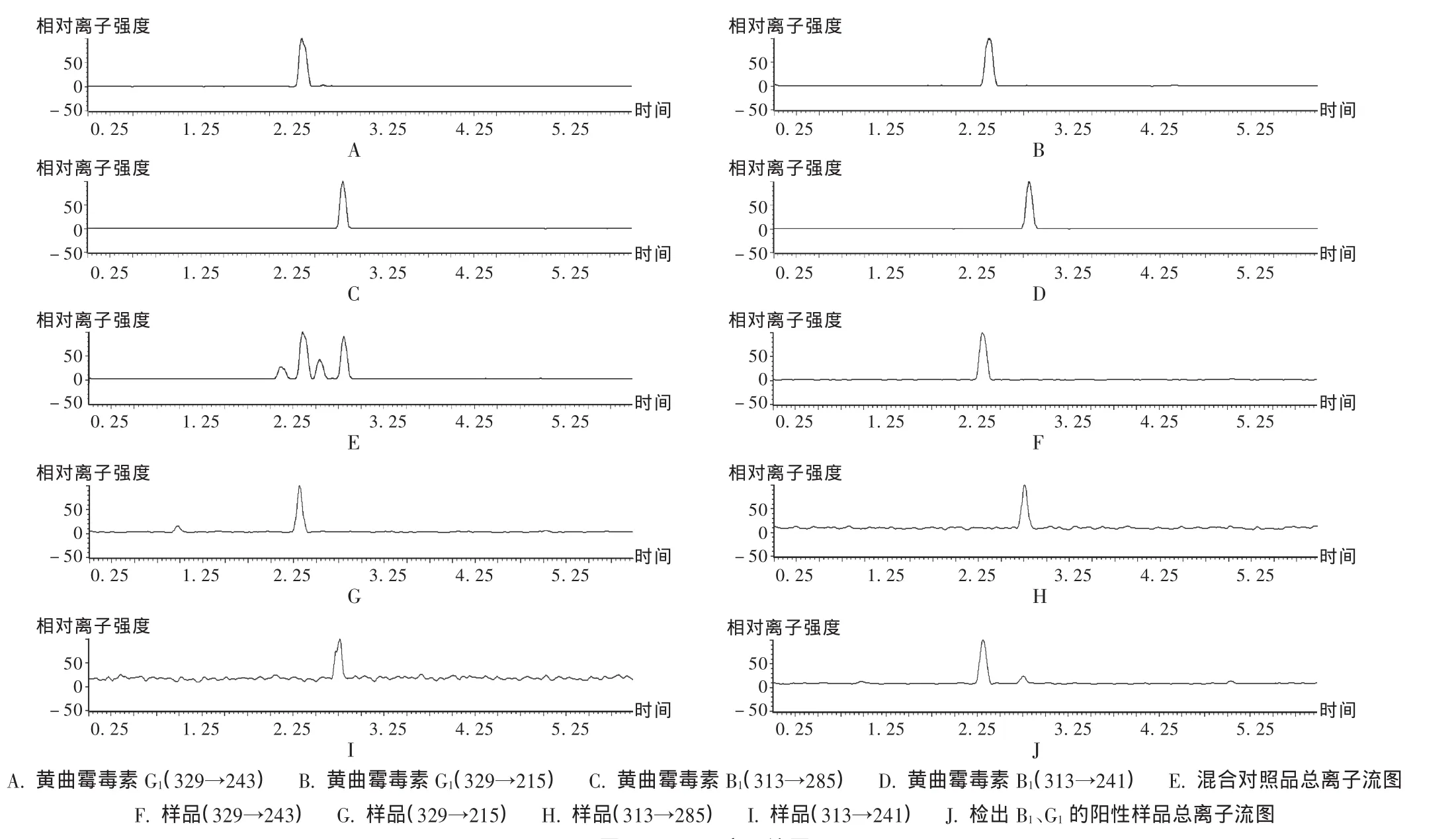
圖3 MRM離子流圖
3 討論
對供試品取樣量、提取方式、稀釋倍數等進行了考察與優化,每一個因素考察均以加樣回收率進行評判,最終確定文中的試驗方法。藥典HPLC方法采用了等度洗脫,洗脫時間長且分離度、峰形均欠佳。本研究改為梯度洗脫,4個成分的保留時間在12.0~17.5 min,峰形明顯改善且分離度均大于1.5。
對UPLC-MS/MS的流動相、梯度洗脫程序、碰撞能量等進行了適當優化,并對數批陽性樣品進行了驗證,由于陽性樣品大多只檢出黃曲霉毒素B1,G1,僅少量檢出B2和G2且量極低,故本試驗最終只對B1,G1進行了確證。
樣品經專屬性極強的免疫親和柱凈化后,在黃曲霉毒素色譜峰附近無干擾峰。鑒于中藥成分復雜,為了排除假陽性干擾,對陽性樣品采取了衍生/不衍生峰面積比及UPLC-MS/MS進行確證。
由于黃曲霉毒素毒性極強而我國目前尚缺乏其毒理學等相關的實驗室數據,因此建議參照2010年版《中國藥典(一部)》陳皮等藥材品種項下規定的黃曲霉毒素殘留量限度,暫訂愈傷靈膠囊中黃曲霉毒素限量為“本品每1 000 g含黃曲霉毒素B1不得過5 μg,含黃曲霉毒素 G2、黃曲霉毒素 G1、黃曲霉毒素 B2、黃曲霉毒素B1的總量不得過10 μg”。本次試驗82批樣品黃曲霉毒素的檢出率為59.76%,不合格率為10.98%。
從愈傷靈膠囊中黃曲霉毒素的檢測情況看來,對于含有容易污染黃曲霉毒素的中藥材的中成藥,尤其是含生藥原粉的制劑,應重點進行黃曲霉毒素監控。
[1]楊曉麗,仇 峰,韋日偉,等.高效液相色譜-串聯質譜法同時測定地龍中4個黃曲霉毒素[J].藥物分析雜志,2012,32(4):627-630.
[2]中華人民共和國香港特別行政區衛生署中醫藥事務部.霉菌毒素(黃曲霉毒素)測定方法[EB/OL].[2005 -09 -01].http://www.cmd.gov.hk/hkcmms/vol1/Docs/Appendix_Ⅶ _Determination_of_Mycotoxins(Aflatoxins)_chi.pdf.
[3]劉 寧,王萌萌,金紅宇,等.高效液相色譜-柱后光化學衍生法測定參苓白術散中黃曲霉毒素 G2、G1、B2、B1[J].中國藥事,2012,26(5):442-445.
[4]鄭 榮,毛 丹,王少敏,等.腦立清丸等6種中成藥中黃曲霉毒素G2、G1、B2、B1的測定[J].中成藥,2010,32(3):418 -422.
[5]劉亞榮,逯雯潔,劉海清,等.免疫親和柱凈化-柱后光化學衍生-高效液相色譜熒光法測定飽和系列制劑中黃曲霉毒素[J].中成藥,2012,34(3):678 - 681.
[6]國家藥典委員會.中華人民共和國藥典(第二增補本)[M].北京:中國醫藥科技出版社,2013:187.
[7]郝愛魚,趙麗元,劉英慧,等.HPLC法測定中藥飲片中黃曲霉毒素殘留量的假陽性研究 [J].藥物分析雜志,2013,33(3):458-464.
