CdS柱撐鈦鈮酸介孔復合材料的制備與光解水制氫活性
2015-06-01 10:45:18蔣少鋒姚倩茹高碧芬陳亦琳林碧洲
無機化學學報 2015年3期
關鍵詞:復合材料
蔣少鋒 姚倩茹 高碧芬 陳亦琳 林碧洲
(華僑大學材料科學與工程學院,廈門361021)
CdS柱撐鈦鈮酸介孔復合材料的制備與光解水制氫活性
蔣少鋒 姚倩茹 高碧芬 陳亦琳 林碧洲*
(華僑大學材料科學與工程學院,廈門361021)
以鈦鈮酸納米片層為主體,CdS納米顆粒為客體,通過剝離-重堆積法合成了不同投料比的CdS-TiNbO5介孔復合材料,材料的層間距為1.19 nm,比表面積約為93 m2·g-1。CdS-TiNbO5復合材料表現出良好的光解水制氫活性,當投料比nCdS∶nHTiNbO5=1∶2時,復合材料在模擬太陽光和可見光下的產氫速率分別為231和184 μmol·h-1·g-1,是CdS的8.6和9.7倍。復合材料光催化活性的提高歸因于比表面積的增加和光生載流子的有效分離。
柱撐材料;鈦鈮酸納米片層;硫化鎘;光催化制氫
氫能是一種綠色環保能源,有望解決因化石燃料過度使用造成的能源危機和環境污染問題[1-2]。半導體光催化分解水制氫可將太陽能有效地轉化為化學能,受到了人們廣泛的關注[2-6]。HTiNbO5是一種層狀半導體氧化物,禁帶寬度為3.44 eV,被廣泛應用于光催化制氫領域[7-9]。但是,寬禁帶的HTiNbO5只能吸收紫外光,太陽能利用率低。為提高鈦鈮酸的光催化性能,人們采用了離子摻雜[10-11]、貴金屬負載[12]和半導體復合[9,13]等方式加以改善。其中,半導體復合能夠促使兩種半導體之間形成異質結結構,以有利于光生電子和空穴的分離,從而提高催化劑的光催化活性。
CdS是一種常見的可見光光催化劑,禁帶寬度為2.4 eV,能與其他半導體復合成具有良好光催化活性的復合材料[14]。研究表明[9,15],HTiNbO5和CdS的導帶底電勢分別為-0.79和-0.81 V(vs NHE),價帶頂分別為2.65和1.61 V。這種能帶位置意味著當兩者復合時,CdS的導帶電子可從轉移到HTiNbO5,而HTiNbO5的價帶空穴則可從轉移到CdS,從而實現光生載流子在不同組分上的空間分離,抑制它們的復合,提高材料的光催化制氫活性。本文以鈦鈮酸納米片層為主體,硫化鎘納米顆粒為客體,采用剝離-重堆積法制備了CdS-TiNbO5介孔柱撐材料,考察了不同CdS含量對復合材料光吸收性質及光催化制氫活性的影響,分析了復合材料光生載流子的分離機理。
1 實驗部分
1.1 催化劑的制備
按文獻[9]方法制得KTiNbO5和HTiNbO5后,將0.5 g鈦鈮酸加入到50 mL的二次水中,用四丁基氫氧化銨(25%,天津市光復精細化工研究所)將懸浮液的pH值調至9~10,攪拌0.5 h,轉移至100 mL水熱釜中,120℃保溫2 h。待水熱釜冷卻,將懸浮液稀釋至500 mL,室溫超聲2 h,離心10 min(6 000 r· min-1),制得TiNbO5-納米片層溶膠。CdS水溶膠參考文獻[16]方法制備。將TiNbO5-納米片層溶膠和CdS水溶膠的濃度均稀釋至10-5mol·L-1。在劇烈攪拌下,將硫化鎘水溶膠緩慢地滴加到TiNbO5-納米片層溶膠中,可以看到淡黃色絮狀物產生。滴加完畢后,繼續在室溫下攪拌12 h,之后置于60℃陳化24 h。待混合液的溫度降至室溫,去除上清液,離心分離,用1∶1(體積比)的乙醇-水混合溶劑清洗產物數次,60℃真空干燥。為了考察CdS的加入量對CdS-TiNbO5復合材料形成和性能的影響,本文制備3種不同投料比的復合材料,CdS和TiNbO5-納米片層的投料物質的量之比為1∶1,1∶2,1∶3的產物分別記為CTN1,CTN2和CTN3。
1.2 催化劑的表征
X射線粉末衍射(XRD)在Rigaku公司生產的SmartLab 3KW衍射儀(Cu Kα,λ=0.154 18 nm)上完成,掃描范圍3°~60°。形貌觀察在Hitachi公司生產的S-4800場發射掃描電子顯微鏡(SEM)和Philips-FEI公司生產的Tecnai F30場發射透射電子顯微鏡(TEM)上進行,X射線電子能譜(EDS)由裝在TEM上的Oxford 7021測量。N2吸附-脫附實驗在Quantachrome公司生產的Nova 1200e全自動比表面和空隙分析儀上完成,測量前樣品在150℃下脫氣2 h,比表面積由BET方法求得。紫外-可見漫反射光譜(UV-Vis DRS)在在Shimadzu UV-2550光譜儀上測定,用BaSO4做基線校正,測試范圍200~800 nm。光電子能譜(XPS)在VGESCALAB MKⅡ型X射線光電子能譜儀上完成,激發源為Al Kα。
1.3 催化劑的電化學測試
光電化學測試在一個三電極體系CHI660B電化學工作站進行,工作電極為涂有樣品的ITO電極,Pt片為對電極,Ag/AgCl為參比電極,0.5 mol·L-1Na2SO4為電解質。其中,交流阻抗測試(EIS)在0.1 V偏壓下進行,頻率范圍為0.1 Hz~100 KHz;光生電流響應測試也在0.1 V偏壓下進行,光源為300 W的氙燈。
工作電極的制備方法如下:先將ITO導電玻璃在丙酮、無水乙醇和二次水中分別超聲30 min后晾干備用。將20 mg樣品加入4 mL二次水中,超聲30 min,然后緩慢滴加到ITO導電玻璃上(有效面積為0.75 cm2),紅外燈烘干。
1.4 光催化分解水制氫活性
光分解水制氫測試是在北京中教金源科技有限公司生產的CEL-SPH2N光催化分解水制氫在線分析系統上進行,光源為300 W氙燈(中教金源CEL-HXF300型)。反應開始之前,先往反應器中加入100 mg催化劑和100 mL含0.001 mol·L-1Na2S與0.001 mol·L-1Na2SO3的混合溶液。反應器安裝完畢后,抽氣至真空,開啟光源,反應開始。反應過程中不斷攪拌溶液,以確保催化劑處于懸浮狀態,與溶液充分接觸,產生的氫氣在Techcomp公司生產的GC7890Ⅱ型氣相色譜儀(譜柱為0.5 nm分子篩)在線測定。
2 結果與討論
2.1 XRD分析
如圖1所示,KTiNbO5的XRD圖與JCPDS 71-1747相符,9.6°強而尖銳的衍射峰為(002)峰。質子化后,該峰移動至10.6°,峰形變寬,結晶度降低。對應的層間距由0.92 nm減少到0.84 nm,這是因為H+替換K+進入到鈦鈮酸鹽層間,使得層間距減小。CdS柱撐到鈦鈮酸層間后,CdS-TiNbO5復合材料的(002)峰向低角度移動至7.4°,相應的層間距擴大到1.19 nm。在CdS-TiNbO5復合材料的XRD中可以觀察到CdS的(111)、(220)和(311)峰(JCPDF10-0454)。這表明CdS納米顆粒已經柱撐到鈦鈮酸層間。同時,復合材料還出現了可歸屬于主體板層的(200)和(020)峰,這說明經剝離和重堆積后主體板層的層結構得到很好保存。不同投料比例的CdS-TiNbO5復合材料具有相似的XRD,只是隨著CdS量的降低,復合材料中CdS的峰強逐漸減弱,這說明不同比例并不影響材料的柱撐結構。

圖1 樣品的XRD圖Fig.1 Powder XRD patterns of samples
2.2 形貌分析
圖2a為前驅物HTiNbO5的SEM圖,可以清晰地看到層板沿著(00l)方向規整堆積。圖2b為CTN2的SEM圖,可以看到,CdS柱撐到鈦鈮酸片層以后,形成了肺泡狀的形貌,這是CdS柱撐材料小晶粒間無序堆積而成。圖2c和d為CTN2的TEM圖,可以看到近于平行的TiNbO5-納米片層條紋,片層有序地堆積在一起,層間有細小顆粒填充,層與層之間的距離為1.2 nm,與XRD的測量結果相符。圖2d白圈局部EDS分析表明,TiNbO5-納米片層與CdS的比例約為2.3∶1,這表明CdS納米顆粒已經成功地柱撐到鈦鈮酸的層間。圖2e示出了CdS的TEM圖及其EDS分析結果。對比圖2e與圖2c和d,可以看出,CTN2的CdS主要存在于TiNbO5-納米片層層間,在材料表面沒有觀察到大量隨機分布的CdS團聚體。

圖2 HTiNbO5(a)和CTN2(b)的SEM圖,CTN2(c和d)和CdS(e)的TEM圖Fig.2 SEM images of HTiNbO5(a)and CTN2(b),TEM image of CTN2(c and d)and CdS(e)
2.3 N2吸附-脫附等溫線
圖3為樣品的氮氣吸附-脫附曲線。由圖可知,HTiNbO5的等溫線沒有出現明顯的滯后環,屬BDDTⅠ型,為無孔材料。CdS柱撐到鈦鈮酸層間后,CdS-TiNbO5復合材料則為Ⅳ類型等溫線,且在p/p0=0.4~1.0之間存在明顯的H3型滯后環,表明復合材料中存在介孔結構,孔呈狹縫狀[9]。如圖3的插圖所示,復合材料的孔徑主要分布在3.1~6 nm間,源于柱撐復合材料小晶粒無序堆積而成的類卡片房結構。

圖3 樣品的氮氣吸附-脫附曲線和CNT2的孔徑分布曲線Fig.3 Nitrogen adsorption(closed symbols)-desorption (opened symbols)isotherms of HTiNbO5,CdS and CTN2
表1為樣品的BET比表面積、孔容及平均孔徑的測量結果。可以看到,KTiNbO5和HTiNbO5的比表面積都比較小,分別是6和13 m2·g-1。CdS和鈦鈮酸片層組裝后,比表面積增加到93 m2·g-1,是前驅物HTiNbO5的7.2倍。而且,相對于HTiNbO5的孔容,復合材料的孔容增加了5.7倍。可見,復合材料的介孔結構對比表面積的提高有很大的貢獻。光催化屬表面控制型過程[2],比表面積的提高有利于提高材料的光催化活性。
2.4 XPS譜

圖4 樣品中S2p(a),O1s(b),Ti2p(c)and Nb3d(d)的XPS譜Fig.4 XPS spectra of S2p(a),O1s(b),Ti2p(c)and Nb3d(d)in HTiNbO5,CdS and CTN2

表1 樣品的平均孔徑、孔容和比表面積Table1 Average pore size,pore volume and specific surface area of samples
圖4為樣品的XPS譜。CdS中S2p3/2和S2p1/2的結合能分別為161.3和162.5 eV,HTiNbO5中Ti2p3/2和2p1/2分別為458.4和464.3 eV,Nb3d3/2和Nb3d1/2分別為206.7和209.5 eV。這些數據均與文獻相符[17-18]。經剝離和重堆積后,所得的復合材料CNT2的S元素的結合能分別為161.1和162.3 eV,較前驅物CdS納米粒子向負方向移動。而CNT2的Ti和Nb元素的結合能則較前驅物HTiNbO5向正方向移動,CNT2的Ti和Nb元素的結合能分別為458.6和464.5 eV,207.0和209.8 eV。結合能數據變化表明,CdS粒子插入主體層板間后,主體Ti4+和Nb5+周圍的電子云密度降低,客體S2-周圍的電子云密度增大,在復合材料中主體鈦鈮酸層板和客體CdS納米顆粒之間形成有效的異質結作用。另一方面,從圖4d可以看出,O1s的XPS峰不對稱,說明存在有不同結合態的氧。利用高斯-洛倫茲峰擬合,HTiNbO5的O1s峰可擬合為530.2和531.7 eV兩個峰。其中,530.2 eV峰可以歸屬為晶格氧,531.7 eV峰對應于表面羥基氧峰[19]。復合材料O1s峰則可擬合為530.3和531.8 eV 2個峰,較HTiNbO5略有增大,這進一步說明主客體存在異質結作用。
2.5 UV-Vis DRS譜
圖5為樣品的紫外-可見漫反射光譜。由圖可知,KTiNbO5和HTiNbO5的起始吸收波長分別為357和364 nm,在可見光區沒有吸收。CdS的起始吸收波長為607 nm,具有可見光光催化活性。相比于鈦鈮酸,復合材料的光吸收范圍紅移至可見光區,CTN1、CTN2和CTN3的起始吸收波長分別為591、574和552 nm。這是因為主客體間形成異質結,兩者發生電子耦合作用,使材料的禁帶寬度減小,可見光響應能力增強,從而有利提高材料光吸收利用率。吸收波長紅移程度隨著CdS投料比的增加而增大,可能與耦合作用的提高有關。

圖5 樣品的紫外-可見漫反射譜Fig.5 UV-Vis diffuse reflectance spectra of samples
2.6 交流阻抗譜和光電流-時間曲線
圖6為樣品電極在0.1 V偏壓300 W氙燈照射下的光電流-時間曲線。HTiNbO5,CdS和CTN2的光電流分別為0.4、1.7和1.9 μA,CTN2的光電流較HTiNbO5和CdS大,說明HTiNbO5與CdS之間的異質結作用使得電子與空穴能在兩組成間迅速轉移,光生載流子的有效分離導致導電玻璃的光生電子增多,光電流強度隨之增加[20]。為了進一步說明HTiNbO5與CdS之間的異質結作用,實驗測量了樣品的交流阻抗譜。如圖7所示,樣品的交流阻抗譜在高頻區都有趨于半圓的圓弧,這部分對應于電子轉移的限制過程[21]。CTN2圓弧的半徑小于HTiNbO5和CdS,說明CTN2內部電子轉移阻力比HTiNbO5和CdS小,電子在CTN2中能更快地轉移到表面,異質結能有效抑制光生載流子的復合。
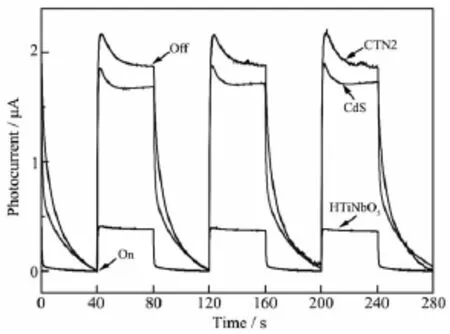
圖6 樣品的光電流-時間曲線Fig.6 Photocurrent-time relationship curves of samples
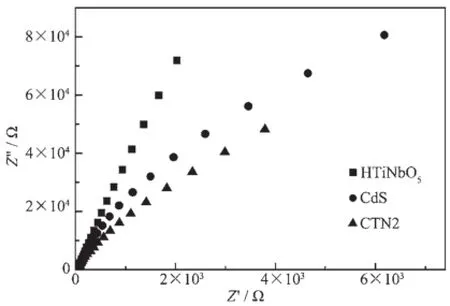
圖7 樣品的交流阻抗譜Fig.7 Nyquist impedance plots of samples
2.7 光解水制氫活性
在300 W氙燈全譜與可見光(λ>420 nm)的照射下,以Na2S/Na2SO3為犧牲劑,不同催化劑催化光解水制氫6 h內產氫平均速率如圖8所示。從圖中可以看出,在氙燈照射下,樣品的光解水制氫活性依次為CTN2>CTN3>CTN1>CdS>P25>HTiNbO5= KTiNbO5。相比于前驅物鈦鈮酸和硫化鎘,復合材料表現出了良好的光催化制氫活性。其中光催化制氫活性最好的為CTN2,產氫速率為231 μmol·h-1·g-1,是CdS納米顆粒(27 μmol·h-1·g-1)的8.6倍。同時,從圖中可以看到,CTN2也具有良好的可見光光催化制氫活性(184 μmol·h-1·g-1),是CdS納米顆粒的9.7倍。CNT1和CNT3的光催化活性較最佳復合比例材料CNT2低,可能與光生載流子分離效率降低有關。CdS-TiNbO5復合材料具有良好光催化性能主要歸因于比表面積的增加和主客體之間耦合作用。此外,為了檢驗催化劑的光催化穩定性,實驗對HTiNbO5,CdS和CTN2進行了3個循環的光催化反應。如圖9所示,CTN2的光催化活性基本保持不變,說明復合材料具有良好的光催化穩定性。
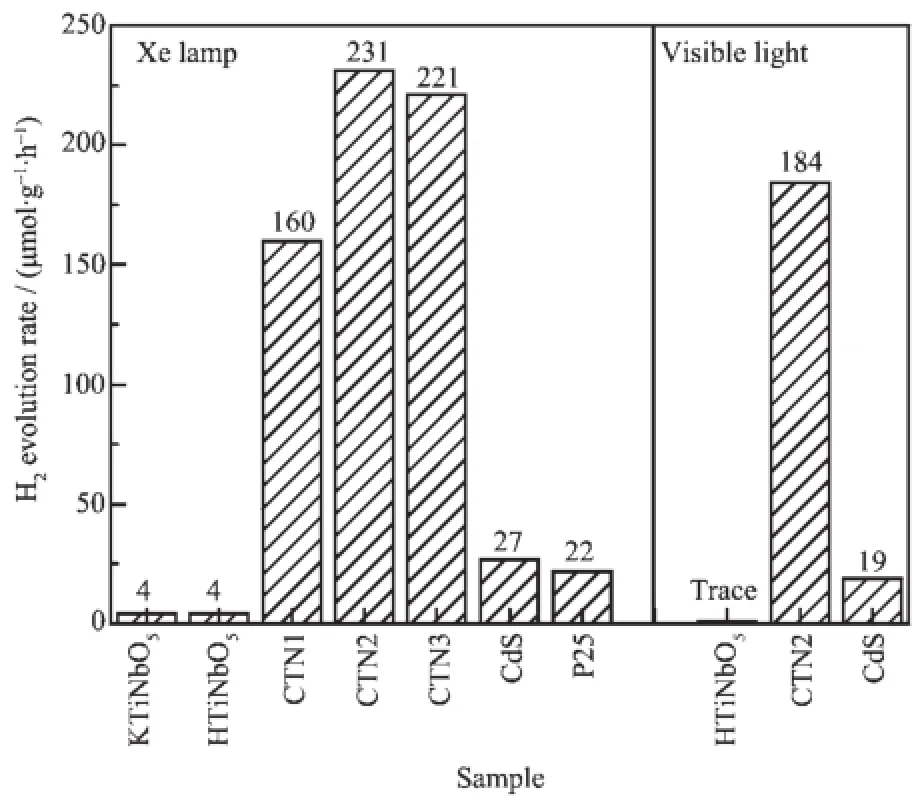
圖8 樣品在300 W氙燈和可見光下的制氫活性Fig.8 H2-evolution photoactivities of samples under irradiation of 300 W xenon lamp and visible light

圖9 樣品的光催化制氫穩定性Fig.9 Photocatalytic stabilities for hydrogen evolution of samples under irradiation of 300 W Xe lamp
HTiNbO5和CdS的導帶底電勢分別為-0.79和-0.81V(vsNHE),價帶頂分別為2.65和1.61V[9,15]。根據組分導帶和價帶的位置可推測復合材料光催化制氫的機理圖如圖10所示。在300 W氙燈的照射下,HTiNbO5和CdS上都會產生光生載流子,電子從價帶躍遷到各自的導帶上。CdS的導帶位置比HTiNbO5更負,電子會從CdS的導帶轉移到HTiNbO5的導帶上,參與還原反應;同時,HTiNbO5的價帶位置比CdS更正,HTiNbO5價帶上產生的空穴會轉移到CdS的價帶上,參與氧化反應。電子和空穴分別轉移到兩個半導體上能夠有效地提高光生載流子的分離效率,從而導致光催化制氫活性的提高。CdS作為一種窄禁帶半導體,與HTiNbO5復合后,窄化了材料的帶隙,使材料吸光范圍向可見光區紅移,賦予復合材料可見光催化活性,提高了太陽能利用率。另外,增強的光催化制氫活性與復合材料的介孔結構也密切相關。介孔結構賦予材料高的比表面積,提供了更多的催化反應活性部位,提高了材料的光催化活性。

圖10 CdS-TiNbO5柱撐復合材料的光催化機理圖Fig.1 0Photocatalytic mechanism of CdS-TiNbO5semiconductor-semiconductor pillared photocatalyst
3 結論
本文分別以鈦鈮酸和CdS為主體和客體,成功合成了不同比例的CdS-TiNbO5柱撐介孔材料。CdS柱撐到鈦鈮酸層間,使得復合材料具有較大的比表面積(~93 m2·g-1)。主體與客體間形成的異質結促使材料的吸收波長紅移至可見光區,并且實現了光生載流子在空間上實現了分離,抑制了電子和空穴的復合,提高了材料催化活性。當投料比nHTiNbO5∶nCdS=2∶1時,在300 W氙燈的照射下,CdS-TiNbO5復合材料光解水制氫效果最好,產氫速率為231μmol·h-1· g-1,是客體CdS的8.6倍;它的可見光活性為184μmol·h-1·g-1,是客體CdS的9.7倍。
[1]Fujishima A.Nature,1972,238:37-38
[2]Chen X B,Shen S H,Guo L J,et al.Chem.Rev.,2010,110: 6503-6570
[3]Maeda K,Domen K.J.Phys.Chem.Lett.,2010,1:2655-2661
[4]Teets T S,Nocera D G.Chem.Commun.,2011,47:9268-9274
[5]Fan W Q,Zhang Q H,Wang Y.Phys.Chem.Chem.Phys., 2013,15:2632-2649
[6]Kudo A,Miseki Y.Chem.Soc.Rev.,2009,38:253-278
[7]Takagaki A,Tagusagawa C,Hayashi S,et al.Energy Environ. Sci.,2010,3:82-93
[8]Inoue K,Suzuki S,Nagai M.J.Electroceram,2008,24:110-114
[9]Fan X R,Lin B Zh,Liu H,et al.Int.J.Hydrogen Energy, 2013,38:832-839
[10]Zhang L H,Hu C H,Zhang J F,et al.Chem.Commun., 2013,49:7507-7509
[11]Zhai Z,Huang Y C,Xu L,et al.Nano Res.,2011,4:635-647
[12]Lin H Y,Chang Y S.Int.J.Hydrogen Energy,2014,39: 3118-3126
[13]Pergentino O,de Brito M M,Andrade H M C,et al.J.Catal., 2014,2014:1-8
[14]Fu J,Chang B B,Tian Y L,et al.J.Mater.Chem.A,2013, 1:3083-3090
[15]Kar A,Kundu S,Patra A.RSC Adv.,2012,2:10222-10230
[16]ZHANG Yu(張宇),ZHANG Jun-Xiang(張俊祥),FU De-Gang(付德剛),et al.Chin.J.Inorg.Mater.(無機材料學報), 1999,15(5):595-600
[17]Zirak M,Akhavan O,Moradlou O,et al.J.Alloys Compd., 2014,590:507-513
[18]Zhai Z,Hu C H,Yang X Y,et al.J.Mater.Chem.,2012, 22:19122-19131
[19]Zhai Z,Yang X Y,Xu L,et al.Nanoscale,2012,4:547-556
[20]Zhu B L,Lin B Z,Zhou Y,et al.J.Mater.Chem.A,2014, 2:3819-3827
[21]Nan Z,Zhang Y H,Pan X Y,et al.J.Phys.Chem.C,2011, 115:23501-23511
Preparation of Mesoporous CdS-Pillared Titanoniobate Nanohybrids and Their Photocatalytic Water-Splitting Activity for Hydrogen Generation
JIANG Shao-FengYAO Qian-RuGAO Bi-FenCHEN Yi-LinLIN Bi-Zhou*
(College of Materials Science and Engineering,Huaqiao University,Xiamen,Fujian 361021,China)
Mesoporous CdS-pillared titanoniobate nanohybrids were successfully prepared via an exfoliationrestacking route.It was found that as-prepared CdS-TiNbO5nanohybrids have an interlayer spacing of 1.19 nm and high specific surface areas of about 93 m2·g-1.The nanohybrids showed a remarkable photocatalytic hydrogen-evolution activity in water splitting.When the molar ratio of CdS and titanoniobate nanosheets was equal to 1∶2,the H2-evolution rate reached 231 and 184 μmol·h-1·g-1under irradiation of 300 W Xe lamp and visible light,which is 8.6 and 9.7 times as high as the blank CdS.The improvement of photocatalytic activity is predominantly ascribed to the enlarged specific surface area and the effective spatial separation of photogenerated carriers between the host and guest.
pillared materials;titanoniobate nanosheets;cadmium sulfide;photocatalytic hydrogen generation
O614.24+2;O643.3
A
1001-4861(2015)03-0514-07
10.11862/CJIC.2015.077
2014-09-29。收修改稿日期:2014-12-03。
國家自然科學基金(No.50872037),福建省自然科學基金(No.2014J01187)資助項目。*
。E-mail:bzlinhqu@126.com,bzlin@hqu.edu.cn
猜你喜歡
建材發展導向(2022年2期)2022-03-08 01:44:04
建材發展導向(2021年14期)2021-08-23 00:56:16
中國材料進展(2019年10期)2019-12-07 05:32:14
纖維復合材料(2018年3期)2018-04-25 07:22:58
電子測試(2017年11期)2017-12-15 08:57:13
山東工業技術(2016年15期)2016-12-01 05:31:34
中國塑料(2015年6期)2015-11-13 03:02:54
中國塑料(2015年11期)2015-10-14 01:14:14
中國塑料(2015年8期)2015-10-14 01:10:41
應用化工(2014年10期)2014-08-16 13:11:29
