綜述原位聚合法制備高性能聚乙烯基復合材料
2015-06-24 14:35:48何富安張黎明
石油學報(石油加工) 2015年2期
何富安, 張黎明
(1.中山大學 化學與化學工程學院 高分子研究所 PCFM和GD HPPC實驗室, 廣東 廣州 510275;2.廣東石油化工學院 化學工程學院高分子材料系, 廣東 茂名 525000)
綜述原位聚合法制備高性能聚乙烯基復合材料
何富安1,2, 張黎明1
(1.中山大學 化學與化學工程學院 高分子研究所 PCFM和GD HPPC實驗室, 廣東 廣州 510275;2.廣東石油化工學院 化學工程學院高分子材料系, 廣東 茂名 525000)
綜述了近年來國內外通過原位聚合法制備高性能聚乙烯基復合材料的研究進展,主要涉及黏土/聚乙烯復合材料、碳納米管/聚乙烯復合材料、石墨類填料/聚乙烯復合材料等體系,重點介紹了相關材料制備時所用負載型催化劑的聚合反應規律,以及所得聚乙烯基復合材料的形貌、結構與性能,還指出了該領域未來研究面臨的一些挑戰。
聚乙烯;復合材料;原位聚合;改性劑
作為五大合成樹脂之一,聚乙烯不僅具有良好的化學穩定性、韌性和電絕緣性,而且具有耐侯、耐低溫、不易吸水等優點。然而,單一聚乙烯材料的尺寸穩定性、耐熱性、耐環境應力開裂性及阻隔性較差,且其韌性有余而剛性和強度不足。因此,采用增強劑并探索有效途徑與方法對聚乙烯進行復合改性,具有重要意義。此外,將聚乙烯與具有光、電、磁等特殊功能的填料復合,還可實現聚乙烯材料的功能化,從而擴展其使用范圍。
迄今為止,制備聚乙烯基復合材料的方法主要有溶液共混、熔融共混和原位聚合[1]。其中,原位聚合方法一般是將配位催化劑首先負載到填料上面,然后以負載催化劑催化乙烯進行聚合反應,最終就地生成聚乙烯基復合材料。與其它2種方法相比,原位聚合法具有以下4個優點[2]。(1)只需經過一步聚合反應就可以在得到聚乙烯的同時得到復合材料;(2)填料分散比較均勻,且與聚乙烯基體之間的界面相互作用較強;(3)能夠制備高填充和超高相對分子質量的聚乙烯基復合材料,這是通過熔融共混和溶液共混都較難實現的,而且高填充聚乙烯基復合材料還可作為母料,進一步與其它聚合物熔融共混制備相應的復合材料;(4)由于使用的是負載催化劑,適用于現有的聚乙烯工業生產設備。因此,原位聚合一直是制備聚乙烯基復合材料的熱門方法。原位聚合制備聚乙烯基復合材料所使用的催化劑主要是Zeigler-Natta催化劑、茂金屬催化劑和后過渡金屬催化劑,所用的填料主要有黏土、碳納米管、石墨類填料、水滑石、二氧化硅、磁性粒子、二氧化鋯等[3-92]。筆者結合自己的相關研究工作,以不同填料改性劑為線索,綜述了近年來國內外通過原位聚合制備高性能聚乙烯基復合材料的最新研究進展。
1 黏土/聚乙烯復合材料
在通過原位聚合制備聚乙烯基復合材料的研究中,黏土/聚乙烯復合材料是研究最廣泛的體系。無機黏土作為層狀硅酸鹽(其典型的2∶1結構如圖1所示),包括了蒙脫土、蛭石、海泡石、凹凸棒等多種類型[1]。無機黏土的片層厚度大約在1 nm左右,由硅、鎂、鋁、氧、氫等元素成,層與層之間有可交換的水合陽離子。黏土自身的結構特點決定了要成功地通過原位聚合法制備黏土/聚乙烯基復合材料,需要考慮催化劑是否能夠負載到黏土片層之間,以及對無機黏土進行有機插層改性兩個關鍵因素。如果催化劑只是負載于黏土的表面,在聚合反應開始后,聚乙烯分子鏈僅能在黏土表面進行包裹,并不能使黏土片層有效剝離,因而不能提高甚至可能會降低材料的性能;如果催化劑能有效地負載于黏土片層之間,那么在聚合過程中,乙烯單體小分子進入到黏土片層之間與催化劑活性中心進行反應,隨著反應的不斷進行,生成的聚乙烯分子鏈不斷增長,當其尺寸增大到超過黏土片層層間距離時,聚合作用力大于黏土片層之間的庫侖引力,從而將黏土片層逐漸推開,最終使其剝離開來并均勻分布于聚乙烯基體之中。另外,無機黏土與有機非極性聚乙烯基體的相容性差,用有機改性劑對無機黏土進行插層改性,既能提高黏土與聚乙烯基體的界面相互作用,又能增大黏土的層間距,使得在聚合過程中乙烯單體小分子更容易進入黏土片層間進行反應。
制備黏土/聚乙烯復合材料的最終目的是為了提高聚乙烯材料的性能或實現其功能化,因此所得復合材料的微觀結構及其對材料綜合性能的影響是科研工作者最關注的問題。與此同時,由于原位聚合法需要將催化劑負載在黏土片層之間催化聚合反應,因此黏土不僅僅是作為納米增強劑的原材料,同時在聚合過程中還為催化劑提供了1個納米級尺寸的聚合反應受限空間。這實質上是個負載催化劑催化乙烯進行聚合的過程,其中必然涉及到負載催化劑的催化性能、各種聚合條件對聚合反應的影響、聚合反應動力學等問題。這些問題同樣是科研工作者所研究的重點。

圖1 2∶1層狀硅酸鹽結構[1]
1.1 黏土負載Zeigler-Natta催化劑體系
黏土負載Zeigler-Natta催化劑分為單組分載體和復合載體2個體系。復合載體主要由黏土和含鎂化合物組成,而其中的含鎂化合物則是工業Zeigler-Natta催化劑的重要載體組分。使用復合載體的目的是為了克服單組分黏土載體對乙烯聚合活性中心的毒害作用,從而維持甚至提高Zeigler-Natta催化劑的活性。
1.1.1 單組分載體體系
Rong等[3-5]對坡縷石納米晶纖維進行高溫處理,以防止黏土所含水分對催化劑的毒害作用,然后利用坡縷石納米晶纖維上的鎂離子空穴負載TiCl4催化劑,再以該負載催化劑催化乙烯聚合,得到坡縷石/聚乙烯復合材料。他們考察了聚合反應溫度和烷基鋁助催化劑類型對該聚合過程的影響,并通過透射電鏡(TEM)證明了坡縷石納米晶纖維均勻分布于聚乙烯基體之中;此外,還發現了聚乙烯的結晶度隨著坡縷石納米晶纖維含量的增加而下降,認為坡縷石納米晶纖維與聚乙烯基體之間強的界面相互作用導致了復合材料力學性能的提高。
Ramazani等[6]為了提高Zeigler-Natta催化劑的負載量,首先對蒙脫土進行酸處理以增加其羥基含量與孔隙率,然后用烷基鋁與酸處理后的蒙脫土反應得到蒙脫土負載鋁氧化合物,最后通過MCl4催化劑(M為Ti或V)與蒙脫土負載鋁氧化合物反應制備負載催化劑,如圖2所示。通過X射線衍射(XRD)和掃描電鏡(SEM)表征,發現蒙脫土以剝離或者是插層形式存在于聚乙烯基體之中,且蒙脫土即使在含量較高的情況下依然能夠分布均勻;熱失重分析(TGA)的結果表明復合材料具有良好的熱穩定性,而差示掃描量熱(DSC)的測試結果則表明復合材料的結晶度大幅下降;此外,蒙脫土納米片層的加入使得復合材料的氣體阻隔性有了顯著提高。
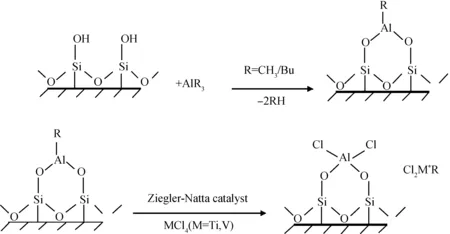
圖2 三異丁基鋁、四氯化鈦、黏土反應原理示意圖[6]
Cui等[7]以蒙脫土負載聚氧酸鈦鹽為催化劑、三異丁基鋁為助催化劑制備了蒙脫土/聚乙烯復合材料。TEM實驗結果證明了蒙脫土片層在聚乙烯基體中能有效剝離和均勻分布;與純聚乙烯相比,復合材料的結晶度有所下降,而熱穩性與力學性能則明顯升高。
Jin等[8]以帶有羥基的有機銨鹽對黏土進行改性,如圖3所示。由于羥基能夠有效地將TiCl4催化劑固定在黏土片層之間,因此可得到黏土片層完全剝離的聚乙烯基復合材料,但該催化體系的催化活性明顯偏低。

圖3 四氯化鈦負載于帶羥基蒙脫土的反應機理[8]
1.1.2 復合載體體系
Yang等[9]將MgCl2與TiCl4依次負載于黏土片層之間,再原位聚合得到了黏土/聚乙烯復合材料,其原理如圖4所示。從圖4可見,利用黏土片層間的羥基來連接MgCl2,再通過Mg—Cl—Ti鍵將催化劑固定在黏土片層之間。XRD和TEM的實驗結果表明,黏土片層在乙烯聚合過程中發生層間剝離,以單片層或數片疊層的形式無規地分散于聚乙烯基體之中;與相對分子質量相近的純聚乙烯相比,極低用量的黏土(質量分數低于1%)就能使復合材料的屈服強度、拉伸強度和拉伸模量有大幅提高。
He等[10]以有機改性黏土-氯化鎂(經四氫呋喃處理)雙組分載體負載TiCl4催化劑,然后以三乙基鋁為助催化劑,通過淤漿聚合得到了有機改性黏土/聚乙烯復合材料。XRD與TEM實驗結果證實,有機改性黏土能在聚乙烯基體中有效剝離并均勻分布,有機黏土的加入能夠有效地提高復合材料的熔融峰溫度、結晶峰溫度、熱穩定性,但卻使其結晶度有所下降。
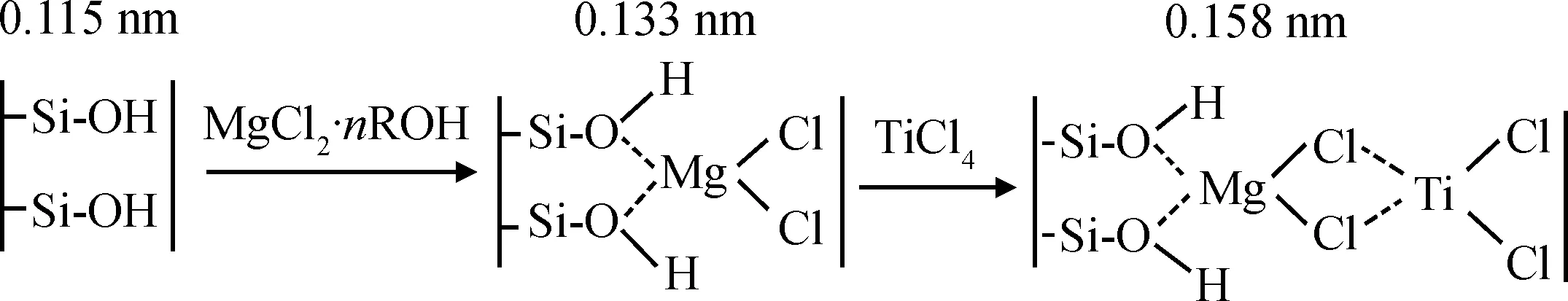
圖4 四氯化鈦催化劑插層負載機理[9]
Nikkhah等[11]以膨潤土-氯化鎂(乙醇鹽型)雙組分載體負載TiCl4催化劑,然后以三異丁基鋁為助催化劑,通過淤漿聚合得到了膨潤土/聚乙烯復合材料。研究表明,膨潤土已經在聚乙烯基體中完全剝離,且僅僅質量分數1%的膨潤土填充量就使得復合材料的氧氣阻隔性能比純聚乙烯提高了200%;雖然復合材料的結晶峰溫度、熱穩定、楊氏模量、拉伸強度都比純聚乙烯有所提高,但韌性卻有所下降。
Cui等[12]以黏土與氯化鎂作為雙組分載體的VOCl3負載催化劑制備了完全剝離型的黏土/聚乙烯復合材料。該催化劑通過氯化鎂提供負載點,避免了VOCl3與黏土的直接接觸,從而具有較高的催化活性。他們發現,黏土片層能有效地剝離;與純聚乙烯相比,復合材料的熔融峰溫度與熱穩定性都有所提高,不同黏土含量的黏土/聚乙烯復合材料的力學強度高出了3.4~7.9 MPa,楊氏模量則提升了23.4%~45.3%。
盡管黏土-氯化鎂雙組分載體能夠維持Zeigler-Natta催化體系的高活性,但由于黏土與氯化鎂的結合效果較差,不能得到均勻的雙組分混合物。而處于游離態的氯化鎂在負載Zeigler-Natta催化劑之后,只能催化乙烯聚合得到不含黏土的純聚乙烯,因此可能會對復合材料的結構造成不良影響。為了克服上述問題,Abedi等[13-15]以丁基辛基鎂替代氯化鎂與蒙脫土復合,制備了混合均勻的雙組分載體,并以該雙組分載體負載TiCl4催化劑,然后在助催化劑三乙基鋁的作用下催化乙烯聚合,得到了蒙脫土/聚乙烯復合材料,其催化劑負載過程如圖5所示。他們考察了影響聚合過程的因素,如聚合反應溫度、乙烯氣體壓強、氫氣鏈轉移劑用量、Al/Ni摩爾比等,發現該負載催化劑具有較好的催化活性,且復合材料的熱穩性能與力學性能都比純聚乙烯有所提高,并存在一個性能最佳的蒙脫土用量。
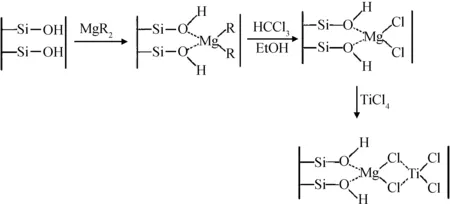
圖5 四氯化鈦催化劑負載過程中可能發生的反應[14]
1.2 黏土負載茂金屬催化劑體系
Huang等[17-19]在有機黏土負載茂金屬催化劑方面進行了一系列工作。他們將茂金屬催化劑Et[Ind]2ZrCl2負載于有機改性黏土,再以MAO為助催化劑催化乙烯與烯醇共聚得到復合材料,其制備路線如圖7所示。由于聚乙烯帶有的羥基與剝離的黏土片層之間有著較強的界面相互作用,使得該復合材料即使經過高溫處理依然顯示出很好的結構穩定性。此外,他們還研究了有機改性黏土類型以及聚合反應溶劑對Et[Ind]2ZrCl2負載催化劑的催化效率和聚乙烯基復合材料結構與性能的影響。
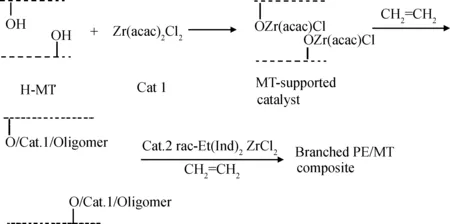
圖6 蒙脫土負載雙功能催化劑制備蒙脫土/聚乙烯復合材料[16]

圖7 通過原位插層共聚方法制備羥基功能化PE/OMMT納米復合材料[17]
Carrero等[20]以海泡石負載(nBuCp)2ZrCl2催化劑制備了海泡石/線性低密度聚乙烯復合材料,并考察了不同負載方式對催化體系活性的影響。他們發現,所得復合材料的形貌結構較好,而且力學性能也有了較大提高。
Wei等[21]使用二氧化硅改性黏土負載茂金屬催化劑Cp2ZrCl2制備得到了黏土-二氧化硅/聚乙烯三元復合材料,制備路線如圖8所示。他們通過TEM觀察到了剝離的黏土片層與二氧化硅納米顆粒分散于聚乙烯基體之中,且聚合所得產物具有使用負載催化劑體系所特有的良好顆粒形態和高堆密度。此外,該黏土-二氧化硅/聚乙烯復合材料還顯示出較好的力學性能。
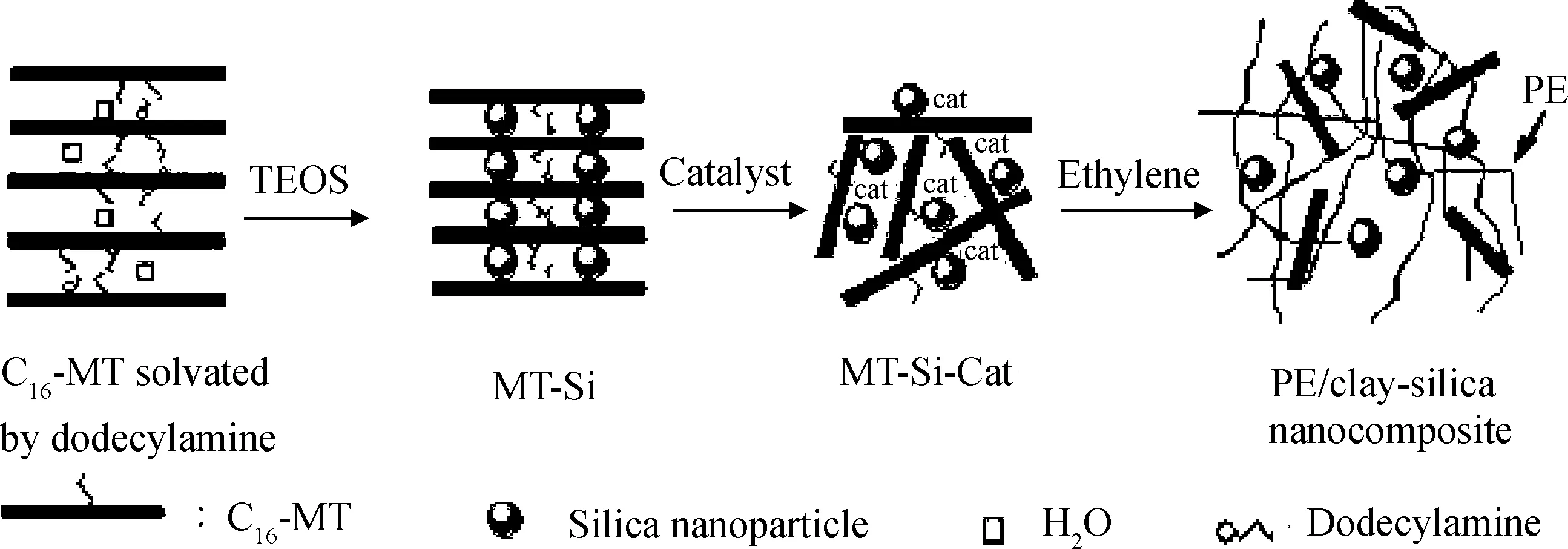
圖8 原位聚合法制備黏土-二氧化硅/聚乙烯三元復合材料路線[21]
Liu等[22-23]使用多種有機改性劑對黏土進行有機改性,然后分別負載Cp2ZrCl2催化劑來催化乙烯均聚以及乙烯與1-辛烯的共聚,制備路線如圖9所示。與Cp2ZrCl2均相催化劑相比,該負載催化劑能夠更為有效地調控聚合產物的相對分子質量、化學組成與結構、堆密度等。

圖9 黏土改性、催化劑的負載以及乙烯的聚合示意圖[22]
Leone等[24]將MAO負載于有機黏土Cloisite 15A,并將其作為Cp2ZrCl2催化劑的助催化劑催化乙烯聚合。該體系的催化活性要高于均相催化劑體系,并且隨著MAO含量的增加而增加;采用元素分析、TGA、TGA-傅里葉轉換紅外光譜(FTIR)、XRD、TEM等手段對被MAO負載的有機黏土表征后發現,MAO與黏土片層之間的有機改性劑發生了反應,破壞了黏土片層的有序結構。他們還對所得復合材料的形貌和熱行為進行了分析研究。
Xalter等[25]將MAO負載于有機改性勃姆石,并將其作為Cp2ZrCl2催化劑的助催化劑催化乙烯聚合。他們發現,以未改性勃姆石和含2%有機改性劑的勃姆石為載體時,催化體系的活性較高,但勃姆石在聚乙烯基體中的分散效果較差;以含20%有機改性劑的勃姆石為載體時,催化體系的活性較低,但勃姆石的分散效果較好。
Maneshi等[26]以三甲基鋁處理不同類型的有機改性黏土后,將其分別作為Cp2ZrCl2催化劑的載體制備負載催化劑,如圖10所示,并研究了該負載催化劑的催化活性以及所得復合材料的形貌。他們發現,有機改性劑的類型對于催化劑的負載效果有著非常重要的影響。由叔胺鹽改性的有機黏土Cloisite 93A具有非常好的催化活性,而其它由季胺鹽改性的有機黏土則催化活性較差,甚至完全沒有活性。

圖10 茂金屬催化劑負載于蒙脫土的可能機理[26]
1.3 黏土負載后過渡金屬催化劑體系
作為早期開創性的工作之一,Heinemann等[27]采用熔融共混和原位聚合兩種方法制備了黏土/高密度聚乙烯復合材料和黏土/線性低密度聚乙烯復合材料,發現使用后過渡金屬催化劑或者茂金屬催化劑進行原位聚合,較之熔融共混方法更有利于黏土片層的剝離分散;通過原位聚合法得到的黏土/聚乙烯復合材料其剛性、強度、韌性、耐熱變形性和阻隔性能都有顯著提高,且材料透明性沒有下降。因此,他們認為該材料具有非常大的應用前景。
He等[28-30]以經過三乙基鋁處理的有機黏土作為載體制備鎳系后過渡金屬負載催化劑,如圖11所示,并研究了Al/Ni摩爾比、聚合反應時間、聚合反應溫度等因素對該負載催化劑聚合催化活性和復合材料黏土填充量的影響。TGA的結果表明,有機黏土的加入能夠明顯地提高該復合材料的熱穩定性。然后,進一步利用了多面齊聚倍半硅氧烷(POSS)的可反應性,通過陽離子交換反應,對無機黏土進行改性,并以該改性黏土作為鎳系后過渡金屬催化劑的載體,再通過原位聚合制備了黏土與POSS協同增強的聚乙烯基復合材料。由于在負載過程中催化劑通過共價鍵固定在黏土的片層之間,因此在聚合反應開始之后,隨著聚乙烯分子鏈在黏土片層之間的不斷增長,黏土片層最終能夠完全剝離,并均勻分布于聚乙烯基體之中。核磁共振碳譜(13C-NMR)的研究結果表明,將該催化劑負載到黏土片層之間后,由于黏土片層納米空間結構的影響,在聚合過程中鎳系后過渡金屬催化劑的鏈行走行為受到了很大限制,從而改變了聚乙烯產物的支鏈分布情況,并顯著降低了聚乙烯產物的支化度,導致復合材料的熔點和結晶度均高于純聚乙烯。此外,黏土片層對催化劑的空間效應作用不僅降低了聚合反應的鏈行走速率,還同時降低了鏈轉移和鏈終止反應的速率,從而增大了聚乙烯產物的相對分子質量。進一步使用連續自成核/退火方法分析了純聚乙烯以及POSS改性黏土/聚乙烯復合材料的聚乙烯基體,結果表明,負載催化劑與均相催化劑所得聚乙烯產物的支化非均勻性存在明顯差異;POSS改性黏土/聚乙烯復合材料的熱穩定性要高于純聚乙烯,當復合材料中POSS改性黏土含量為質量分數9.75%時,其50%的熱失重溫度要比純聚乙烯高62℃。動態機械熱分析(DMA)的研究結果表明,與純聚乙烯相比,POSS改性黏土/聚乙烯復合材料的儲存模量有了大幅提高,當POSS改性黏土質量分數為9.75%時,其在20℃的儲存模量提高了1226%。
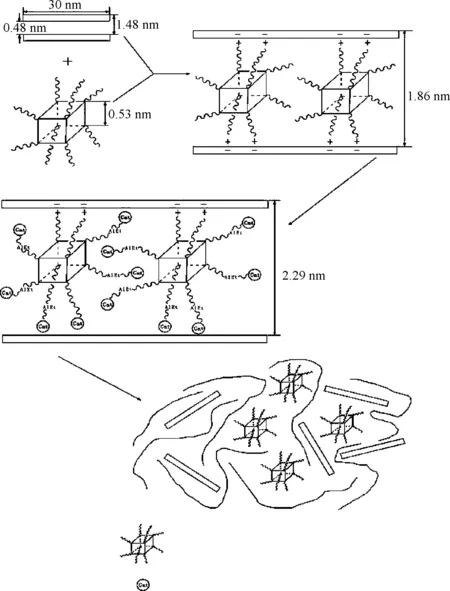
圖11 通過原位聚合制備黏土與POSS協同增強的聚乙烯基復合材料[29]
Shin[31]通過多步反應,如圖12所示,將乙烯基反應基團連接到黏土片層之間,然后再負載鎳系化合物得到后過渡金屬負載催化劑。在聚合過程中,乙烯單體進入到黏土片層之間進行聚合,使得黏土片層有效剝離,并且生成的聚乙烯分子鏈通過共價鍵與黏土片層相連,增加了黏土與聚乙烯基體之間的界面相互作用。
Ray等[32]將鐵系后過渡金屬催化劑負載于經過MAO預先處理的蒙脫土后得到負載催化劑,并將其用于催化乙烯聚合。他們發現,與以未經MAO處理蒙脫土作為催化劑載體的體系相比,該方法能更有效地使黏土片層剝離;負載催化劑的活性與Al/Fe摩爾比無關,而聚乙烯基體的結晶度和晶粒尺寸則會受到蒙脫土剝離程度的影響。
Mignoni等[33]以有機改性蒙脫土為載體,通過鎳系后過渡金屬-MAO(或三甲基鋁)體系催化乙烯聚合,結果表明,催化劑活性與黏土填充量可以通過改變Al/Ni摩爾比等因素進行有效調控;黏土含量的改變對復合材料的性能有著明顯的影響;由于蒙脫土納米片層有效剝離并均勻分布于聚乙烯基體之中,提高了復合材料的力學性能、結晶峰溫度、熔融峰溫度。
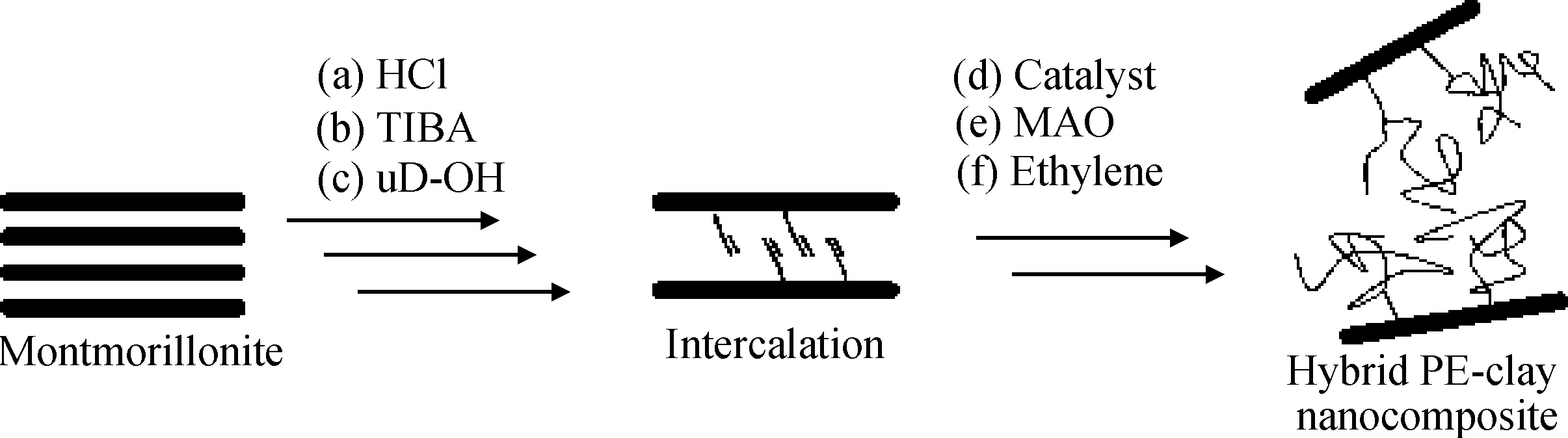
圖12 通過雙功能有機改性劑對黏土進行改性并原位聚合制備共價鍵連接的黏土/聚乙烯復合材料[31]
通常,對黏土進行有機改性用到的都是有機小分子,有可能對催化劑活性和復合材料性能造成不良影響。Scott等[34]針對這一問題,以經過酸處理的蒙脫土負載鎳系后過渡金屬催化劑催化乙烯聚合反應,如圖13所示。他們認為,催化劑配體上的亞甲基和蒙脫土片層上的鋁元素存在著路易斯酸堿相互作用,可以在不使用有機改性的情況下,使催化劑直接負載到黏土的片層之間。實驗結果表明,該催化體系的活性較高,并且能夠實現黏土片層的有效剝離。
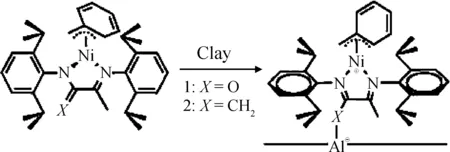
圖13 黏土活化鎳系后過渡金屬催化劑的可能機理[34]
Leone等[35]將鐵系后過渡金屬催化劑負載于經MAO改性的黏土后得到負載催化劑,并將其催化乙烯聚合得到復合材料。他們發現,(1)由于催化劑被負載之后活性中心的分散性較好,表現出比均相催化劑更高的活性;(2)由于黏土載體的保護作用,負載催化劑比均相催化劑具有更長的反應壽命;(3)由于黏土載體能夠降低鏈轉移的速率,使得產物的相對分子質量增加。
2 碳納米管/聚乙烯復合材料
碳納米管由于其獨特的納米結構和優良的電、磁、光、力學和熱傳導等性能,引起了人們廣泛重視。將碳納米管作為填料與聚合物共混,對提高聚合物性能以及實現材料的功能化具有重要意義。然而,由于碳納米管之間存在范德華力作用并且極易纏繞團聚,要將碳納米管應用在聚合物基體中并充分發揮其優異性能,具有相當的難度[93]。制備碳納米管復合材料的兩種主要方法是共混法和原位聚合法。與共混法相比,原位聚合法獲得的復合材料中碳納米管分散更均勻,并且碳納米管與聚合物基體之間具有更強的相互作用[36]。
2.1 碳納米管負載Zeigler-Natta催化劑體系
Ramazani等[37-38]采用以二乙氧基鎂和帶羥基多壁碳納米管作為雙組分載體的TiCl4負載催化劑,制備了多壁碳納米管/超高分子量聚乙烯,其催化劑負載機理如圖14所示。研究結果表明,碳納米管在復合材料中的分散效果良好;碳納米管能夠明顯提高復合材料的楊氏模量、屈服應力和斷裂拉伸強度。他們還采用TGA表征了該復合材料的熱穩定性,并采用Friedman、Ozawa、Flynn、Wall和Kissinger等動力學方法研究了復合材料的熱失重行為,發現碳納米管的加入能夠增加材料的熱分解活化能,從而提高了復合材料的熱穩定性。
Park等[39]采用TiCl4-EtAl3催化體系制備了碳納米管/超高分子量聚乙烯復合材料。摩擦實驗的結果表明,與純超高分子量聚乙烯和熔融共混制備的碳納米管/超高分子量聚乙烯復合材料相比,原位聚合制備的碳納米管/超高分子量聚乙烯復合材料具有更好的耐磨性。他們認為其原因主要是,(1)原位聚合制備的碳納米管/超高分子量聚乙烯復合材料其碳納米分散均勻,與聚乙烯基體界面作用力較強;(2)原位聚合制備的碳納米管/超高分子量聚乙烯復合材料其熱傳導系數更高,能夠及時地將摩擦過程中產生的熱量傳遞出去,降低摩擦表面的溫度,減少了摩擦的損耗。
Liu等[40]先用格氏試劑處理帶羧基、羥基等功能基團的多壁碳納米管,然后負載TiCl4催化劑,再以AlEt3為助催化劑催化乙烯聚合,如圖15所示,制備得到了具有“芯-皮”結構的多壁碳納米管/聚乙烯復合材料,并研究了該材料的結晶行為、熱穩定性、介電性能。
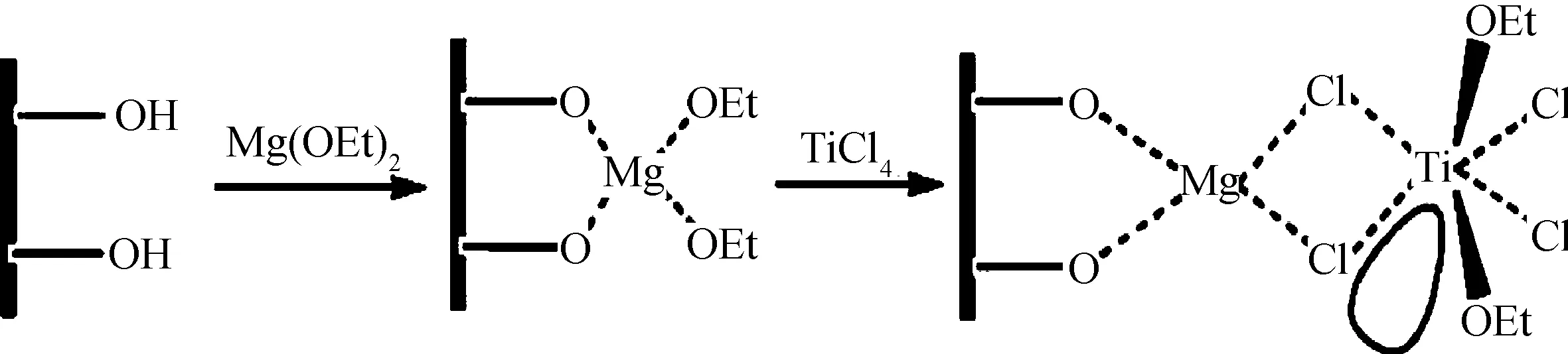
圖14 以二乙氧基鎂和帶羥基多壁碳納米管負載四氯化鈦催化劑的機理[37]
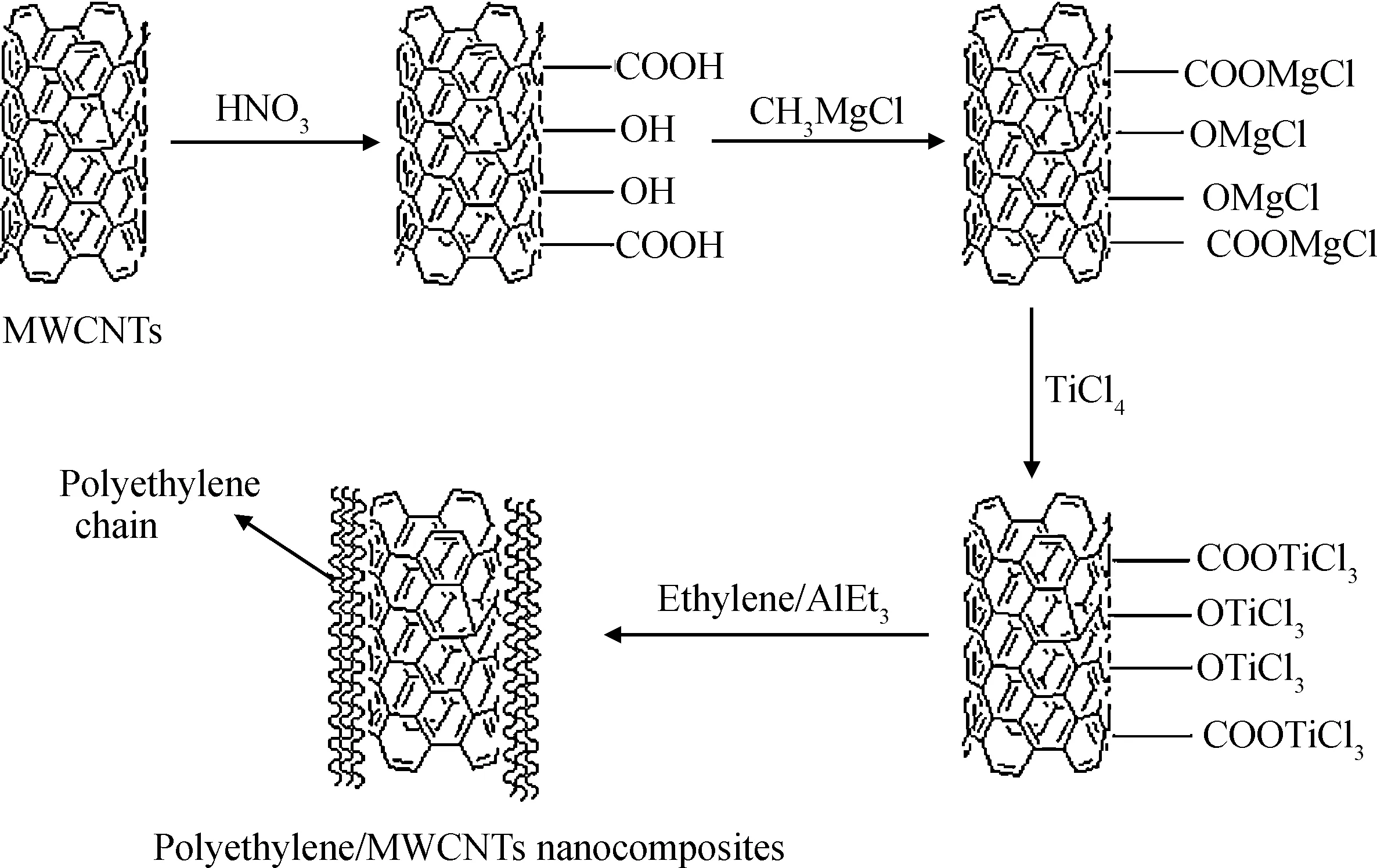
圖15 多壁碳納米管/聚乙烯復合材料制備過程示意圖[40
2.2 碳納米管負載茂金屬催化劑體系
茂金屬負載催化劑體系是通過原位聚合制備碳納米管/聚乙烯復合材料的最常用催化體系。通過該體系制備復合材料的一般過程如圖16所示[41-43]。其中步驟(i)與步驟(ii)不是必須步驟。碳納米管表面經過處理后引入了羧基、羥基等功能基團得到功能化碳納米管,功能基團的引入有利于MAO和催化劑的負載;將MAO負載于功能化碳納米管表面,可以避免雜質對催化劑的毒害作用;在碳納米管上負載催化劑,在助催化劑作用下,催化乙烯聚合得到復合材料。某些碳納米管負載茂金屬催化劑體系甚至可以表現出比均相催化劑更高的活性[42-43],其原因可能是催化劑在負載后分散得更好,不像均相催化劑那樣團聚在一起,從而能夠使得催化劑活性中心與乙烯單體小分子充分接觸,并避免了雙分子失活現象。
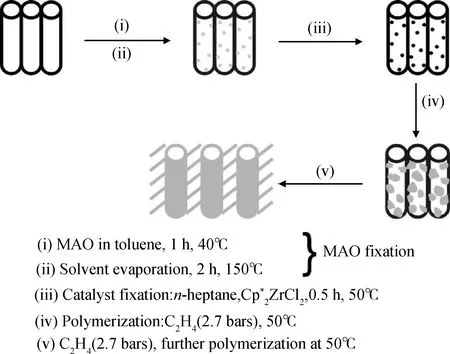
圖16 茂金屬催化劑負載過程以及其催化乙烯聚合示意圖[41]
Tang等[44]首先經過多步反應將乙烯基團接枝到多壁碳納米管表面,然后在rac-(en)(THInd)2ZrCl2-MAO催化體系作用下,催化改性碳納米管與乙烯共聚合得到多壁碳納米管/聚乙烯復合材料,如圖17所示。1H-NMR、SEM、FTIR、TGA等表征結果證明了聚乙烯成功地接枝到了碳納米管表面。
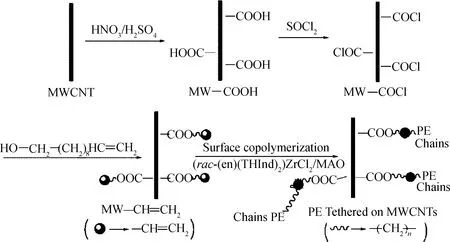
圖17 多壁碳納米管/聚乙烯復合材料制備過程示意圖[44]
Kim等[45-47]則采用一鍋法制備了多壁碳納米管/聚乙烯復合材料。先依次用三異丁基鋁與MAO對甲苯溶劑中的多壁碳納米管進行處理,然后直接向溶液中加入rac-Et(Ind)2ZrCl2催化劑,再通入乙烯單體進行反應,得到復合材料。他們發現,碳納米管的加入能夠有效地提高復合材料的熱傳導系數、電導率、復合黏度、儲存模量、損耗模量、沖擊強度等性能。他們還進一步研究了該復合材料的熱穩定性和非等溫結晶動力學過程。
Bahuleyan等[48]采用了一種相對比較簡單的一鍋法制備了多壁碳納米管/聚乙烯復合材料。首先在甲苯溶劑中加入Cp2TiCl2催化劑(或Cp2ZrCl2催化劑)和碳納米管,然后對其進行超聲波處理,最后加入助催化劑并通入乙烯氣體進行聚合反應。他們發現,通過選擇不同類型的催化劑和聚合反應條件,可以控制復合材料的外觀形貌,得到類似于“香腸”形貌或者是串晶形貌的復合材料。
Dong等[49-50]以經MAO處理的多壁碳納米管作為載體負載Cp2ZrCl2催化劑催化乙烯聚合。在該聚合過程中(T=50℃,[Al]/[Zr]=1000),生成的聚乙烯分子鏈以高長/徑比的碳納米管作為模板不斷進行包裹,最終得到了纖維狀形貌的復合材料;當[Al]/[Zr]摩爾比和聚合溫度繼續升高時,纖維狀形貌將向無定型形貌轉變。他們進一步發現,分別以末端封閉碳納米和末端開口碳納米管作為載體時,可以分別得到纖維狀和無定型兩種不同的聚乙烯形貌。他們認為,以開口碳納米管作為載體時,催化劑可以進入碳納米管的內部,聚合反應開始之后隨著聚乙烯分子鏈在碳納米管內部不斷地增長,最終導致碳納米分裂成為碎片;后續生成的聚乙烯將以不規則的碳納米管碎片作為模板不斷包裹,從而得到了具有不規則形貌的聚乙烯,其機理如圖18所示。Trujillo等[51]以類似的方法分別制備了以單壁碳納米管、雙壁碳納米管、多壁碳納米管為填料的聚乙烯基復合材料,測定了反應終止后助催化劑的殘留物三氧化二鋁在復合材料中的含量,同時還考察了復合材料的形貌結構、晶體成核過程、結晶行為等,發現各種類型的碳納米管對聚乙烯都有著很好的結晶成核作用。
Toti等[52]用改性甲基鋁氧烷(MMAO)對多壁碳納米管進行預處理,然后負載雙組分催化劑,得到催化劑體系1 CoCl2N2Th+[(η5-C5Me4)SiMe2(NtBu)]-TiCl2和催化劑體系2 CoCl2N2Th+Cp2ZrCl2,前者為乙烯齊聚催化劑,后者為乙烯共聚催化劑,再催化單一乙烯單體進行聚合,制備了多壁碳納米管/線性低密度聚乙烯復合材料;通過改變催化劑組分的比例和共聚合反應的條件可以有效地控制聚乙烯基體的支化度以及接枝鏈類型。

圖18 Dong等所制備的無規聚乙烯的形成過程[49]
Park等[53-54]采用一鍋法制備了多壁碳納米管/高密度聚乙烯復合材料。在反應釜中依次加入正庚烷、MAO、多壁碳納米管與Cp2ZrCl2催化劑的正庚烷淤漿混合物后,再通入乙烯氣體進行聚合反應。力學測試的結果表明,多壁碳納米管的加入使得復合材料的楊氏模量與純聚乙烯相比提高了359%。他們進一步的研究發現,碳納米管可以與帶正電的二茂鋯活性中心形成配位鍵,并充當具有強給電子能力的配體;鈦的親電活性可以使得碳納米管和半茂金屬鈦(Cp*TiCl3,Cp*:五甲基環戊二烯基)有強烈的電子相互作用,從而影響附著在碳納米管表面的半茂鈦催化劑的催化性能;Cp*TiCl3可以通過Cp*與多壁碳納米管表面的相互作用從而吸附于碳納米管壁,形成碳納米管-Cp*TiCl3負載催化劑;以該催化體系催化乙烯聚合得到的復合材料,其多壁碳納米被超高分子量聚乙烯包裹后直徑為30~70 nm,大于碳納米管直徑10~15 nm。
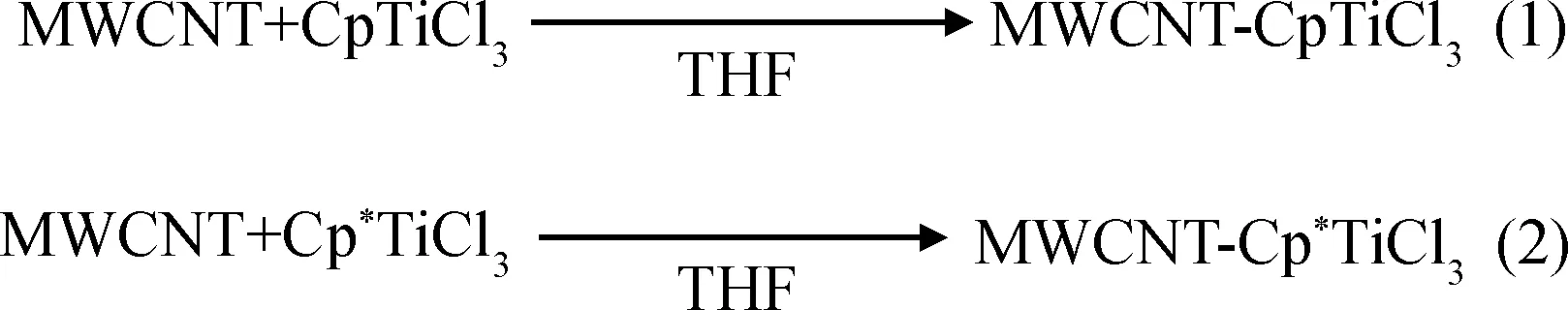
圖19 Park等制備的無規聚乙烯的形成過程[54]
2.3 碳納米管負載后過渡金屬催化劑體系
采用后過渡金屬催化劑體系制備碳納米管/聚乙烯復合材料的報道相對較少。Zhang等[55]制備了含有大體積配體的高活性鐵系后過渡金屬催化劑,如圖20所示。該鐵系催化劑能夠通過π-π相互作用負載到多壁碳納米管的表面,且催化活性要好于均相催化劑。研究表明,多壁碳納米管能夠均勻分散在聚乙烯基體之中。

圖20 鐵系后過渡金屬催化劑負載于多壁碳納米管的示意圖[55]
2.4 母料共混體系
原位聚合法的優點在于碳納米管分散比較均勻,與聚乙烯基體之間的界面作用較強,且能夠制備高填充碳納米管/聚乙烯復合材料。因此不少科研工作者以含有高碳納米管填充量的聚乙烯基復合材料作為母料與聚合物熔融共混制備相應的復合材料。
何富安[56]采用母料熔融共混(mPEC)與直接熔融共混(dPEC)2種方法制備了多壁碳納米管/聚乙烯復合材料。在mPEC制備過程中,首先通過TiCl4-三乙基鋁催化體系原位聚合制備多壁碳納米管填充量為質量分數53%的聚乙烯基復合材料,如圖21所示,然后以其作為母料與高密度聚乙烯樹脂熔融共混。研究結果表明,mPEC的力學性能要優于dPEC,說明在原位聚合過程中生成的聚乙烯包裹層對多壁碳納米管的分散以及界面改性起到了非常重要的作用;多壁碳納米管質量分數為3%的dPEC3和mPEC3的拉伸屈服強度分別為25.23 MPa和27.86 MPa,比純聚乙烯提高了25.2%和38.3%,楊氏模量分別為963 MPa和1081 MPa,比純聚乙烯提高了26.2%和41.7%;碳納米管質量分數為1%的dPEC1和mPEC1的彎曲強度為29.50 MPa和31.55 MPa,分別比純聚乙烯提高了16.3%和24.4%,彎曲模量為753 MPa和883 MPa,分別比純聚乙烯提高了21.8%和42.9%。與之相類似,Tong[36]等的研究結果則表明,采用母料熔融共混法得到的單壁碳納米管(質量分數1%)/聚乙烯復合材料的屈服強度、拉伸強度、拉伸模量、斷裂伸長率、斷裂能分別比直接熔融共混法得到的復合材料提高了25%、15.2%、25.4%、21%、38%。此外,Vega等[57-58]先以多壁碳納米管作為載體負載Cp2ZrCl2催化劑,然后催化乙烯聚合得到母料,再與高密度聚乙烯熔融共混得到復合材料,并研究了該復合材料的熔融流變行為、結晶行為、力學性能等。
Bredeau等[59]采用以碳納米管負載rac-Et(Ind)2ZrCl2-MAO催化劑體系催化乙烯與降冰片烯共聚,得到共聚物基復合材料,并以該共聚物基復合材料作為母料采用熔融共混法制備了碳納米管/乙烯-醋酸乙烯共聚物復合材料。力學性能測試表明,碳納米管的加入能夠大幅度提高復合材料的楊氏模量。此外,他們還在碳納米管上依次負載MMAO和Cp2*TiCl2催化劑,然后催化乙烯聚合制備得到母料,再與乙烯-醋酸乙烯共聚物熔融共混得到復合材料。他們的研究表明,母料熔融共混法可以比直接熔融共混法得到更好的碳納米管分散效果,從而有助于提高復合材料的熱穩定性、力學性能、導電性能。
為了避免由于使用MAO、烷基鋁等助催化劑而導致聚乙烯產物中含有大量的氧化鋁殘余物,Ravasio等[60]以[Sc(g5-C5Me4SiMe3)(g1-CH2SiMe3)2(THF)]作為催化劑,[Ph3C][B(C6F5)4]為助催化劑,少量三異丁基鋁作為清除劑,制備了多壁碳納米管/聚乙烯-降冰片烯共聚物復合材料,如圖22所示。他們發現,在以該復合材料為母料與TOPAS環烯烴類共聚物進行熔融共混得到的復合材料中,碳納米管分散均勻。
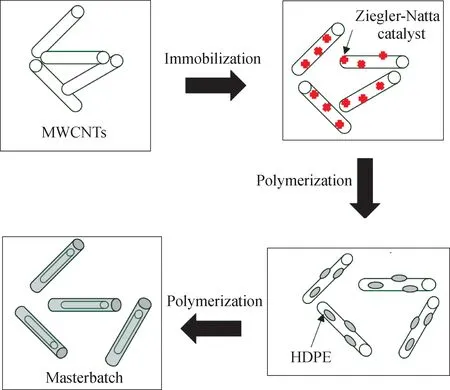
圖21 多壁碳納米管/聚乙烯母料的制備過程[56]
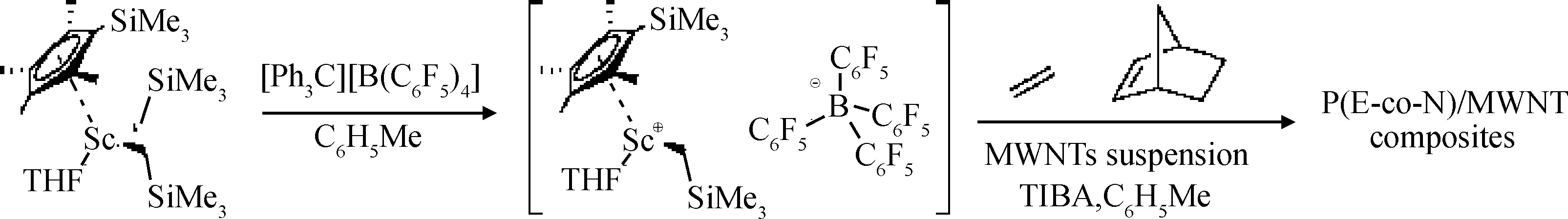
圖22 多壁碳納米管/聚乙烯-降冰片烯共聚物復合材料母料的制備過程[60]
3 石墨類填料/聚乙烯復合材料
石墨類填料包括普通石墨、氧化石墨、納米石墨片、石墨烯等。其中的石墨烯是近幾年繼富勒烯、碳納米管后發展起來的又一種新型碳納米材料,在化學、物理、材料、電子等領域都掀起了研究熱潮。石墨烯具有優異的導電性和傳熱性、超高的模量、高的長/徑比(長度和厚度的比值)和極大的比表面積,是理想的增強劑。
Alexandre等[61-62]采用3種不同方法制備了石墨/聚乙烯復合材料。一種方法是將催化劑負載于經過MAO改性的石墨,然后進行原位聚合;另一種方法是一鍋法,即將石墨加入溶劑中,再依次添加助催化劑MAO與催化劑進行原位聚合;第3種方法即是機械共混法。他們還研究了不同方法制備得到的石墨/聚乙烯復合材料的外貌形態、熱性能、導電性能等。
Fabiana等[63-64]以Cp2ZrCl2為催化劑,MAO為助催化劑,納米石墨片為填料,制備了納米石墨片/聚乙烯復合材料。在催化過程中,聚乙烯分子鏈不斷對其插層,使得石墨納米片層不斷剝離,并摻雜進聚乙烯基體均勻分散,由于納米石墨片的存在降低了Cp2ZrCl2催化劑的活性。他們進一步發現,納米石墨片的加入能夠使復合材料的熱失重起始溫度和剝離溫度分別提高30℃和5℃,并且存儲模量和電導率也有所上升。
Hu等[65]用經過有機硅氧烷處理的氧化石墨烯先后負載MAO與Cp2ZrCl2,并用該負載催化體系制備了聚乙烯基復合材料。由于有機基團被引入到石墨烯片層之間,使得石墨烯片層在聚合過程中能夠有效剝離,并均勻分散在聚乙烯基體之中,其制備過程如圖23所示。使用非有機改性氧化石墨烯負載催化劑得到的復合材料則出現了明顯的相分離。此外,氧化石墨烯/聚乙烯復合材料的熔點比采用均相催化劑得到的純聚乙烯有明顯提高。
Lee等[66]在多層石墨烯上負載了(n-BuCp)2ZrCl2催化劑后,在助催化劑MAO的作用下催化乙烯聚合物,同樣發現了聚乙烯分子鏈對多層石墨烯的插層剝離現象,而且石墨烯在聚合過程中起到了形貌模板的作用。石墨烯負載(n-BuCp)2ZrCl2催化劑的過程如圖24所示。TEM的結果表明,生成的復合材料具有類似于菊花花瓣的形貌。此外,石墨烯還起到了催化劑大配體的作用,使得到的聚乙烯比均相催化劑具有更高的相對分子質量和多分散系數。
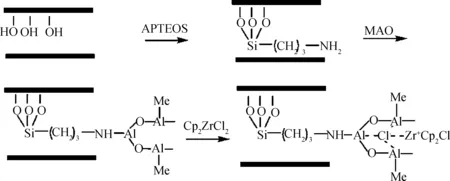
圖23 茂金屬催化劑負載于氧化石墨烯的過程[65]

圖24 (n-BuCp)2ZrCl2通過π-π作用負載于多層石墨烯的示意圖[66]
Todd等[67]在較低溫度(40℃)和壓力(乙烯氣體壓強為1.01×105Pa)下,以熱還原石墨烯-(n-BuCp)2ZrCl2)-MAO催化體系制備了聚乙烯基復合材料。結果表明,當熱還原石墨烯質量分數為5.2%時,復合材料的拉伸強度和楊氏模量比純聚乙烯分別提高了57%和170%。
Stürzel等[68]以帶羥基的功能化石墨烯為載體,采用鉻系催化劑和MAO助催化劑在高壓反應釜中原位制備了石墨烯/超高分子量聚乙烯復合材料,并與其它碳系填料負載催化劑體系以及勃姆石負載催化劑作比較,負載過程如圖25所示。由于石墨烯帶有的羥基能夠很好地使負載催化劑分散在正庚烷中,因此該催化體系表現出最好的催化活性和形貌控制能力;當功能化石墨烯的質量分數僅為1%時,就能夠大幅度提高復合材料的剛性、斷裂伸長率和結晶能力。
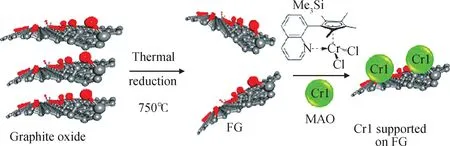
圖25 鉻系催化劑負載于功能石墨烯的過程[68]
Choi等[69]則以氮摻雜還原氧化石墨烯作為Cp2ZrCl2與Cp2TiCl2催化劑的載體,制備多壁碳納米管/超高分子量聚乙烯復合材料。他們發現,在聚合物過程中氮摻雜石墨烯起到了相當于配體的作用,能夠調節金屬中心空間和電子屬性,所以得到的聚乙烯比使用均相催化劑得到的聚乙烯具有更高的相對分子質量,其制備過程如圖26所示。
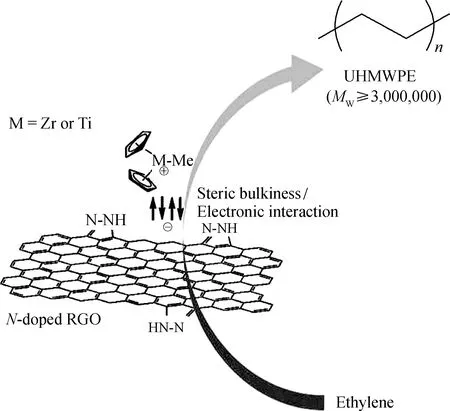
圖26 茂金屬催化劑負載于氮摻雜還原氧化石墨烯以及制備多壁碳納米管/超高分子量聚乙烯復合材料[69]
4 原位聚合制備其它聚乙烯基復合材料
除了上述3種主要填料之外,科研工作者還將原位聚合法應用于制備其它聚乙烯基復合材料,在提高聚乙烯材料的性能以及實現聚乙烯功能化方面都取得了相當的成果。
作為早期開創性的工作之一,Alexandre[70-71]等使用了不同的催化劑載體,包括玻璃珠、石墨、氫氧化鎂、高嶺土等等,負載茂金屬催化劑,并通過原位聚合制備了多種無機填料/聚乙烯復合材料。
He等[72-73]以經過有機改性的水滑石作為載體負載鎳系后過渡金屬催化劑后,再通過原位聚合法制備了水滑石/聚乙烯復合材料,如圖27所示。考察了各種聚合條件,包括聚合反應時間、聚合反應溫度和Al/Ni摩爾比對負載催化劑活性的影響,結果表明,(1)隨著反應時間的延長,負載催化劑的活性逐漸下降;(2)隨著聚合溫度的升高,負載催化劑催化活性先增大后減小,在30~40℃范圍內達到最大值;(3)隨著Al/Ni摩爾比的增大,負載催化劑的活性也增大,Al/Ni摩爾比在300~900之間時,活性的增長速率較快,當Al/Ni摩爾比達到1200后催化劑活性增長的速率放緩。他們進一步研究了負載催化劑與非負載均相催化劑在相同聚合條件下的聚合反應動力學,結果表明,對催化劑進行負載后,提高了催化劑的活性中心的長期穩定性;在負載催化劑體系中,水滑石對生成的聚乙烯具有很好的模板作用,得到了顆粒狀外貌的聚乙烯產物,而使用均相催化劑得到的則是無定型結構聚乙烯;水滑石片層對于負載在片層之間的催化劑存在著受限空間效應,它降低了鏈轉移和鏈終止反應的速率,從而增大了聚乙烯產物的相對分子質量;水滑石片層以納米級的尺寸均勻分散于聚乙烯的基體之中,得到了部分剝離型的水滑石/聚乙烯復合材料。由于聚乙烯與水滑石納米片層之間有著較強的界面作用,從而限制了聚乙烯分子鏈的運動,并減弱了聚乙烯的結晶能力,因此隨著水滑石含量的增加,聚乙烯的結晶度下降;水滑石/聚乙烯復合材料的熱穩定性要高于純聚乙烯,當水滑石質量分數為10.32%時,其30%的熱失重溫度要比純聚乙烯高61℃。此外,水滑石/聚乙烯復合材料在熔融狀態下的儲能模量、損耗模量、復合黏度均高于純聚乙烯。
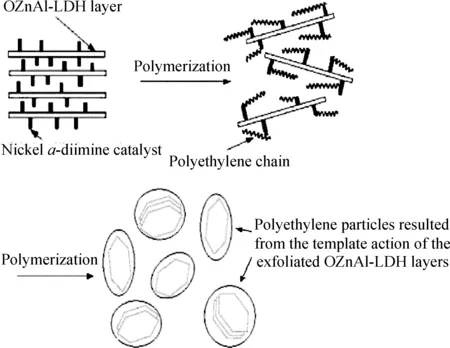
圖27 鎳系后過渡金屬催化劑負載于有機改性水滑石以及制備水滑石/聚乙烯復合材料[73]
Monteil等[74]為了增加二氧化硅納米粒子與聚乙烯基體之間的相容性,首先在二氧化硅納米粒子表面接枝辛烯基團或者是辛基硅氧烷,然后通過鎳系后過渡金屬催化劑原位聚合,得到二氧化硅/聚乙烯復合材料。他們發現,通過改變鎳系催化劑的配體可以有效地調控二氧化硅的分布形態。
Wang等[75]將Cp2ZrCl2催化劑在MAO的作用下負載于高嶺土,如圖28所示,然后通過原位聚合得到了高嶺土/聚乙烯復合材料。溶劑抽提實驗和FTIR的結果顯示,聚乙烯基體與高嶺土粒子之間存在著較強的界面相互作用;DMA的結果則顯示,這種界面作用能夠有效地阻礙聚乙烯高分子鏈的運動。此外,與機械共混相比,原位聚合使得高嶺土具有更好的分散性。
Zhang等[76]將Ziegler-Natta催化劑負載于納米鐵電材料(Pb,Sr)TiO3(PST)粉末上,然后再通過控制聚合反應時間,得到不同PST含量的PST/聚乙烯復合材料。結果表明,PST能夠均勻地分散于聚乙烯基體之中,并且PST含量對PST/聚乙烯復合材料的介電常數有著很大影響。
Jongsomjit等[77-80]在原位聚合制備線性低密度聚乙烯基復合材料方面進行了大量工作。他們通過二氧化硅和二氧化鋯納米粒子負載rac-Et(Ind)2ZrCl2-MAO催化劑體系催化乙烯與α-烯烴共聚,分別得到了二氧化硅/線性低密度聚乙烯復合材料和二氧化鋯/線性低密度聚乙烯復合材料。當使用二氧化硅作為載體時,催化劑的活性較低,但當使用二氧化鋯作為載體時,催化劑活性卻有大幅度提高,為二氧化硅負載體系的5倍。他們認為,這可能是由于二氧化硅納米粒子與MAO之間較強的結合力造成了空間位阻,使得乙烯單體難以接近催化劑活性中心。根據TEM的結果,可以看到二氧化硅與二氧化鋯納米粒子在線性低密度聚乙烯基體中都有較好的分散效果。他們還比較了不同粒徑(10 nm和15 nm)的二氧化硅作為載體時對催化劑活性的影響,發現當二氧化硅粒徑較大時,與MAO之間的相互作用較弱,催化劑具有更高的活性,并且使得α-烯烴接觸催化劑活性中心的幾率增加,如圖29所示,從而導致聚乙烯的支化度上升和熔點下降。此外,他們還通過類似的原位聚合方法制備了三氧化二鋁/線性低密度聚乙烯復合材料、氧化鋅/線性低密度聚乙烯復合材料和二氧化鈦/線性低密度聚乙烯復合材料,并研究了它們的結構與性能[81-83]。

圖28 Cp2ZrCl2/MAO負載于高嶺土表面形成活性中心[75]
Cheng等[84]通過母料熔融共混法制備了二氧化硅/聚乙烯復合材料。結果表明,母料熔融共混得到的復合材料中二氧化硅的分散效果要比直接熔融共混得到的復合材料更好;在二氧化硅質量分數為2.0%時,母料熔融共混得到的復合材料的最高熱失重速率溫度、結晶度和拉伸強度分別為472℃、68.3%和28.3 MPa,而直接熔融共混得到的復合材料其相應的數值則為430℃,66.3%和26.3 MPa。
Covarrubias等[85]采用原位聚合和熔融共混兩種方法制備了多孔層狀磷酸鋁/聚乙烯復合材料。他們發現,盡管原位聚合法可以使得多孔層狀磷酸鋁充分剝離,但其結構卻遭到了一定程度的破壞,而熔融共混則能夠較好地保持多孔層狀磷酸鋁的結構。小分子氣體H2/CO的滲透測試結果則表明,采用熔融共混得到的多孔層狀磷酸鋁/聚乙烯復合材料的透氣性能要好于采用原位聚合法得到的復合材料。

圖29 不同尺寸的納米SiO2對乙烯聚合的影響[77]
Wang等[86]以MCM-41分子篩與MgCl2為復合載體,TiCl4為催化劑制備了MCM-41/聚乙烯母料,然后與聚乙烯樹脂熔融共混,得到了相應的復合材料。與純聚乙烯相比,所得MCM-41(質量分數1%)/聚乙烯復合材料的力學性能有了大幅提高,其拉伸模量、拉伸強度和斷裂伸長率分別提高了24.7%、28.3%、23.5%。
Kaleel等[87]以Cp2ZrCl2和Cp2TiCl2為催化劑、MAO為助催化劑、質量分數1%錳摻雜的二氧化鈦為填料制備了相應的聚乙烯基復合材料,考察了填料含量、聚合反應溫度、乙烯氣體壓強等因素對復合材料熱性能與力學性能的影響。結果表明,使用Cp2ZrCl2催化劑對聚乙烯的熔點影響不大,并能使復合材料的斷裂強度、楊氏模量、斷裂伸長率有明顯提高。
Wang等[88-90]將TiCl4催化劑負載在磁性納米粒子表面制備得到了磁性納米催化劑,然后通過催化乙烯原位聚合得到新型磁性聚乙烯復合材料,其制備路線如圖30所示。他們研究了該負載催化劑的催化活性,同時還使用不同烷基鋁對磁性粒子進行改性,發現三乙基鋁的改性效果最好。江洪流等[91]則用納米四氧化三鐵磁性粉末作為載體,負載α-二亞胺鎳催化劑制備得到磁性納米催化劑,通過乙烯在納米催化劑粒子表面原位聚合得到新型磁性支化聚乙烯復合材料,結果表明,四氧化三鐵較均勻地分散在聚乙烯基體中,其尺寸在20 nm左右。

圖30 磁性催化劑與磁性聚乙烯復合材料的制備路線[88]
Park等[92]分別采用機械共混和二氧化鋯負載TiCl4催化劑催化乙烯原位聚合2種方法制備了二氧化鋯/超高分子量聚乙烯復合材料。采用原位聚合方法得到的復合材料的二氧化鋯納米粒子分散得更為均勻,并且與聚乙烯基體有著更強的界面作用,因此力學性能與耐磨性都比使用機械共混方法得到的復合材料有了顯著提高。
5 結束語
原位聚合是迄今用于制備填料/聚乙烯復合材料的最有效方法之一,該方法為提高聚乙烯材料的性能以及實現其功能化提供了重要途徑。然而,目前該領域研究仍面臨以下一些挑戰:
(1) 如何使負載催化劑具有與均相催化劑相當、甚至更高的催化活性;
(2) 如何使聚合后得到的聚乙烯基復合材料粉末具有好的顆粒形貌和高的堆密度;
(3) 如何使填料能夠在聚乙烯基體中穩定分散,不至于復合材料在加工與使用過程中出現相分離現象;
(4) 如何提高聚乙烯基復合材料的綜合性能以及有效地實現其功能化。
因此,深入研究聚乙烯基復合材料的原位聚合反應規律及其影響因素,同時探索有關催化劑效率、產物形貌控制、復合材料結構與性能之間的相互關系,對于高性能聚乙烯基復合材料的研究和工業化生產將具有十分重要的意義。
[1] RAY S S, OKAMOTO M. Polymer/layered silicate nanocomposites: A review from preparation to processing[J]. Progress in Polymer Science, 2003, 28(11): 1539-1641.
[2] ABEDI S, ABDOUSS M. A review of clay-supported Ziegler-Natta catalysts for production of polyolefin/clay nanocomposites through in situ polymerization[J]. Applied Catalysis A General, 2014, 475(11): 386-409.
[3] RONG J F, LI H Q, JING Z H, et al. Novel organic/inorganic nanocomposite of polyethylene. I. preparation via in situ polymerization approach[J]. Journal of Applied Polymer Science, 2001, 82(8): 1829-1837.
[4] RONG J F, JING Z H, LI H Q, et al. A polyethylene nanocomposite prepared via in-situ polymerization[J]. Macromolecular Rapid Communications, 2001, 22(5): 329-334.
[5] RONG J F, SHENG M, Li H Q, et al. Polyethylene-palygorskite nanocomposite prepared via in situ coordinated polymerization[J]. Polymer Composites 2002, 23(4): 658-665.
[6] RAMAZANI A S A, TAVAKOLZADEH M, BANIASADI H, et al. In situ polymerization of polyethylene/clay nanocomposites using a novel clay-supported Ziegler-Natta catalyst[J]. Polymer Composites 2002, 30(10): 1388-1393.
[7] CUI L Q, CHO H Y, SHIN J W, et al. Polyethylene-montmorillonite nanocomposites: Preparation, characterization and properties[J]. Macromolecular Symposia 2007, 260: 49-57.
[8] JIN Y H, PARK H J, IM S S, et al. Polyethylene/clay nanocomposite by in-situ exfoliation of montmorillonite during Ziegler-Natta polymerization of ethylene[J]. Macromolecular Rapid Communications 2002, 23(2): 135-140.
[9] YANG F, ZHANG X Q, ZHAO H C, et al. Preparation and properties of polyethylene/ montmorillonite nanocomposites by in situ polymerization[J]. Journal of Applied Polymer Science, 2003, 89(13): 3680-3684.
[10] HE F A, ZHANG L M, YANG F, et al. Polyethylene nanocomposites obtained from in-situ polymerization using supported Ziegler-Natta catalyst system[J]. Journal of Macromolecular Science Part A Pure and Applied Chemistry, 2003, 44(1): 11-15.
[11] NIKKHAH S J, RAMAZANI S A A, BANIASADI H, et al. Investigation of properties of polyethylene/clay nanocomposites prepared by new in situ Ziegler-Natta catalyst[J]. Materials & Design, 2009, 30(7): 2309-2315.
[12] CUI L Q, WOO S I, et al. Preparation and characterization of polyethylene(PE)/clay nanocomposites by in situ polymerization with vanadium-based intercalation catalyst[J]. Materials & Design, 2009, 61(4): 453-460.
[13] ABEDI S, ABDOUSS M, NEKKOMANESH-HAGHIFHI M, et al. New clay-supported ZieglerNatta catalyst for preparation of PE/clay nanocomposites via insitu polymerization[J]. Journal of Applied Polymer Science, 2013, 128(3): 1879-1884.
[14] ABEDI S, ABDOUSS M, NEKKOMANESH-HAGHIFHI M,et al. PE/clay nanocomposites produced via in situ polymerization by highly active clay-supported Ziegler-Natta catalyst[J].Polymer Bulletin, 2013, 70(4): 1313-1325.
[15] ABEDI S, ABDOUSS M, NEKKOMANESH-HAGHIFHI M, et al. Highly exfoliated PE/Na+MMT nanocomposite produced via in situ polymerization by a catalystsupported on a novel modified Na+MMT[J]. Polymer Bulletin, 2013, 70(10): 2783-2792.
[16] WANG J, LIU Z Y, GUO C Y, et al. Preparation of a PE/MT composite by copolymerization of ethylene with in-situ produced ethylene oligomers under a dual functional catalyst system intercalated into MT layer[J]. Macromolecular Rapid Communications, 2001, 22(17): 1422-1426.
[17] HUANG Y J, YANG K F, DONG J Y, et al. Copolymerization of ethylene and 10-undecen-1-ol using a montmorillonite-intercalated metallocene catalyst: Synthesis of polyethylene/montmorillonite nanocomposites with enhanced structural stability[J]. Macromolecular Rapid Communications, 2006, 27(15): 1278-1283.
[18] HUANG Y J, QIN Y W, DONG J Y, et al. PE/OMMT nanocomposites prepared by in situ polymerization approach: Effects of OMMT-intercalated catalysts and silicate modifications[J]. Journal of Applied Polymer Science, 2012, 123(5): 3106-3166.
[19] HUANG Y J, QIN Y W, DONG J Y, et al. PE/OMMT nanocomposites catalyzed by the OMMT Intercalated Et[Ind]2ZrCl2in slurry polymerization: Effects of organic solvent on ethylene polymerization behaviors and the nanocomposite structures[J]. Journal of Applied Polymer Science, 2011, 119(1): 190-200.
[20] CARRERO A, VAN GRIEKEN R, SUAREA I, et al. Development of a new synthetic method based on in situ strategies for polyethylene/clay composites[J]. Journal of Applied Polymer Science, 2012, 126(3): 987-997.
[21] WEI L M, TANG T, HUANG B T, et al. Development of a new synthetic method based on in situ strategies for polyethylene/clay composites[J]. Journal of Polymer Science Part A-Polymer Chemistry, 2004, 42(4): 941-949.
[22] LIU C B, TANG T, WANG D, et al. In situ ethylene homopolymerization and copolymerization catalyzed by zirconocene catalysts entrapped inside functionalized montmorillonite[J]. Journal of Polymer Science Part A-Polymer Chemistry, 2003, 41(14): 2187-2192.
[23] LIU C B, TANG T, HUANG B T, et al. Zirconocene catalyst well spaced inside modified montmorillonite for ethylene polymerization: Role of pretreatment and modification of montmorillonite in tailoring polymer properties[J]. Journal of Catalysis, 2004, 221(1): 162-169.
[24] LEONE G, BERTINI F, CANETTI M, et al. In situ polymerization of ethylene using metallocene catalysts: Effect of clay pretreatment on the properties of highly filled polyethylene nanocomposites[J]. Journal of Polymer Science Part A-Polymer Chemistry, 2008, 46(16): 5390-5403.
[25] XALTER R, HALBACH TS, MULHAUPT M, et al. New polyolefin nanocomposites and catalyst supports based on organophilic boehmites[J]. Macromolecular Symposia, 2006, 236: 145-150.
[26] MANESHI A, SOARES J B P, SIMON L C, et al. Polyethylene/clay nanocomposites made with metallocenes supported on different organoclays[J]. Macromolecular Chemistry and Physics, 2011, 212: 216-228.
[27] HEINWMANN J, REICHERT P, THOMANN R, et al. Polyolefin nanocomposites formed by melt compounding and transition metal catalyzed ethene homo- and copolymerization in the presence of layered silicates[J]. Macromolecular Rapid Communications, 1999, 20(8): 423-430.
[28] HE F A, ZHANG L M, JIANG H L, et al. A new strategy to prepare polyethylene nanocomposites by using a late-transition-metal catalyst supported on AlEt3-activated organoclay[J]. Macromolecular Rapid Communications, 2007, 67(7-8): 1727-1733.
[29] HE F A, ZHANG L M. Using inorganic POSS-modified laponite clay to support a nickel alpha-diimine catalyst for in situ formation of high performance polyethylene nanocomposites[J]. Nanotechnology, 2007, 17(24): 1727-1733.
[30] HE F A, ZHANG L M. Study on branching structure, melting, and crystallization of polyethylene prepared by nickel a-diimine catalyst covalently intercalated inside OapPOSS-modified laponite clay gallery[J]. Polymer Testing, 2014, 35(24): 80-86.
[31] SHIN S Y A, SIMON L C, SOARES J B P, et al. Polyethylene-clay hybrid nanocomposites: In situ polymerization using bifunctional organic modifiers[J]. Polymer, 2003, 44(18): 5317-5321.
[32] RAY S, GALGALI G, SIVARAM S, et al. In situ polymerization of ethylene with bis(imino)pyridine iron(II) catalysts supported on clay: The synthesis and characterization of polyethylene-clay nanocomposites[J]. Journal of Polymer Science Part A-Polymer Chemistry, 2005, 43(2): 304-318.
[33] MIGNONI M L, MAERINS S J V, DE SOUZA M O, et al. Polyethylene-montmorillonite nanocomposites obtained by in situ polymerization of ethylene with nickel-diimine catalysts[J]. Journal of Applied Polymer Science, 2011, 122(3): 2159-2165.
[34] SCOTT S L, PEOPLES B C, YUNG C, et al. Highly dispersed clay-polyolefin nanocomposites free of compatibilizers, via the in situ polymerization ofα-olefins by clay-supported catalysts[J]. Chemical Communicatoins, 2008, (35): 4186-4188.
[35] LEONE G, BERTINI F, CANETTI M, et al. Long-lived layered silicates-immobilized 2,6-bis(imino)pyridyl iron (II) catalysts for hybrid polyethylene nanocomposites by in situ polymerization: effect of aryl ligand and silicate modification[J]. Journal of Polymer Science Part A-Polymer Chemistry, 2009, 47(2): 548-564.
[36] TONG X, LIU C, CHENG H M, et al. Surface modification of single-walled carbon nanotubes with polyethylene via in situ Ziegler-Natta polymerization[J]. Journal of Applied Polymer Science, 2004, 92(6): 3697-3700.
[37] AMOLI B M, RAMAZANI S A, AHMAD L H, et al. Surface modification of single-walled carbon nanotubes with polyethylene via in situ Ziegler-Natta polymerization[J]. Journal of Applied Polymer Science[J]. 2012, 125(S1): E453-E456.
[38] JAFAR S, RAMAZANI S A, FARHAD K, et al. Thermal degradation behavior and kinetic analysis of ultra high molecular weight polyethylene based multi-walled carbon nanotube nanocomposites prepared via in-situ polymerization[J]. Journal of Macromolecular Science Part A Pure and Applied Chemistry, 2012, 49(9): 749-757.
[39] PARK H J, KIM J H, SEO Y S, et al. Wear behavior of in situ polymerized carbon nanotube/ultra high molecular weight polyethylene composites[J]. Macromolecular Research, 2013, 21(9): 965-970.
[40] LIU Z, YU M S, WANG J, et al. Preparation and characterization of novel polyethylene/carbon nanotubes nanocomposites with core-shell structure[J]. Journal of Industrial and Engineering Chemistry, 2014, 20(4): 1804-1811.
[41] BONDUEL D, MAIANIL M L, ALEXANDRE M, et al. Supported coordination polymerization: A unique way to potent polyolefin carbon nanotube nanocomposites[J]. Chemical Communications, 2005, (6): 781-783.
[42] BONDUEL D, BREDEAU S, ALEXANDRE M, et al. Supported metallocene catalysis as an efficient tool for the preparation of polyethylene/caron nanotube nanocomposites:Effect of the catalytic system on the coating morphology[J]. Journal of Materials Chemistry, 2007, 17(22): 2359-2366.
[43] LI S Y, CHEN H, CUI D M, et al. Structure and properties of multi-walled carbon nanotubes/polyethylene nanocomposites synthesized by in situ polymerization with supported Cp2ZrCl2catalyst[J]. Polymer Composites, 2010, 31(3): 507-515.
[44] TANG J J, LI S Y, WANG Y H, et al. In situ ethylene copolymerization with an olefin-type monomer for one-pot synthesis of polyethylene tethered on multi-walled carbon nanotubes[J]. Journal of Industrial and Engineering Chemistry, 2013, 31(10): 1329-1333.
[45] KIM J H, SEO Y S, HONG S M, et al. Preparation of PE/MWNT nanocomposites by in-situ metallocene polymerization[J]. International Journal of Material Forming[J], 2009, 2(1): 873-875.
[46] KIM J H, HONG S M, KWAK S J, et al. Physical properties of nanocomposites prepared by in situ polymerization of high-density polyethylene on multiwalled carbon nanotubes[J]. Physical Chemistry Chemical Physics, 2013, 11(46): 10851-10859.
[47] KIM J H, KWAK S J, HONG S M, et al. Nonisothermal crystallization behaviors of nanocomposites prepared by in situ polymerization of high-density polyethylene on multiwalled carbon nanotubes[J]. Macromolecules, 2010, 43(24): 10545-10533.
[48] BAHULEYAN B K, ATIEH M A, DE S K, et al. Easy one-pot method to control the morphology of polyethylene/carbon nanotube nanocomposites using metallocene catalysts[J]. Journal of Polymer Research, 2012, 19(2): 9744.
[49] DONG X C, WANG L, SUN T X, et al. Study on ethylene polymerization catalyzed by Cp2ZrCl2/carbon nanotube system[J]. Journal of Molecular Catalysis A-Chemical, 2006, 255(1-2): 10-15.
[50] DONG X C, WANG L, DONG L B, et al. Preparation of nano-polyethylene fibres using Cp2ZrCl2/carbon nanotube catalytic system[J]. Materials Letters, 2007, 61(14-15): 3111-3115.
[51] TRUJILLO M, AENAL M L,MULLER A J, et al. Thermal and morphological characterization of nanocomposites prepared by in-situ polymerization of high-density polyethylene on carbon nanotubes[J]. Macromolecules, 2007, 40(17): 6268-6276.
[52] TOTI A, GIAMBASTIANI G,BIANCHINI C, et al. Tandem action of early-late transition metal catalysts for the surface coating of multiwalled carbon nanotubes with linear low-density polyethylene[J]. Chemistry of Materials, 2007, 20(9): 3092-3098.
[53] PARK S J, YOON S W,CHOI H C, et al. Pristine multiwalled carbon nanotube/polyethylene nanocomposites by immobilized catalysts[J]. Chemistry of Materials, 2008, 20(14): 4588-4594.
[54] PARK S J, CHOI I S. Production of ultrahigh-molecular-weight polyethylene/pristine MWCNT composites by half-titanocene catalysts[J]. Advanced Materials, 2009, 21(8): 902-905.
[55] ZHANG L P, ZHANG W J, SERP P, et al. Ethylene polymerization catalyzed by pyrene-tagged iron complexes: the positive effect ofπ-conjugation and immobilization on multiwalled carbon nanotubes[J]. Chem Cat Chem, 2014, 6(8): 1310-1316.
[56] 何富安. 新型聚乙烯納米復合材料的制備與性能研究[D].廣州:中山大學化學與化學工程學院. 2007.
[57] VEGA J F, MARTINEZ-SALAZAR J, TRUJILLO M, et al. Rheology, processing, tensile properties, and crystallization of polyethylene/carbon nanotube nanomposites[J]. Macromolecules, 2009, 42(13): 4719-4272.
[58] VEGA J F, SILVA D Y, VIXWNRW-AKUQUE E, et al. Influence of chain branching and molecular weight on melt rheology and crystallization of polyethylene/carbon nanotube nanocomposites[J]. Macromolecules, 2014, 47(16): 5668-5681.
[59] BREDEAU S, BOGGIONI L, BERTINI F, et al. Ethylene-norbornene copolymerization by carbon nanotube-supported metallocene catalysis: generation of high-performance polyolefinic nanocomposites[J]. Macromolecular Rapid Communications, 2007, 28(7): 822-827.
[60] RAVASIO A, BOGGIONI L, TRITTO I, et al. A non-PFT (polymerization filling technique) approach to poly(ethylene-co-norbornene)/MWNTs nanocomposites by in situ copolymerization with scandium half-sandwich catalyst[J]. Journal of Polymer Science Part A-Polymer Chemistry, 2009, 47(21): 5709-5719.
[61] ALEXANDRE M, PLUTA M, DUBOIS P, et al. Metallocene catalyzed polymerization of ethylene in the presence of graphite, 1-Synthesis and characterization of the composites[J]. Macromolecular Chemistry and Physics, 2001, 202(11): 2239-2246.
[62] PLUTA M, ALEXANDRE M, BLACHER S, et al. Metallocene-catalyzed polymerization of ethylene in the presence of graphite II Structure and electrical properties of the composites[J]. Polymer, 2001, 42(22): 9293-9300.
[63] FABIANA D C F, JONATHAN M G, NaRA S B, et al. Polyethylene/graphite nanocomposites obtained by in situ polymerization[J]. Journal of Polymer Science Part A-Polymer Chemistry, 2010, 48(3): 692-698.
[64] FABIANA D C F, NARA S B, ANA P G, et al. Thermal, electrical, and mechanical properties of polyethylene-graphene nanocomposites obtained by in situ polymerization[J]. Journal of Applied Polymer Science . 2012, 128(5): 2630-2637.
[65] HU Z, LIU C B. Polyethylene/graphite oxide nanocomposites obtained by in situ polymerization using modified graphite oxide-supported metallocene catalysts[J]. Journal of Polymer Research. 2013, 20(1): 39.
[66] LEE J S, KO Y S. Synthesis of petaloid graphene/polyethylene composite nanosheet produced by ethylene polymerization with metallocene catalyst adsorbed on multilayer graphene[J]. Catalysis Today. 2014, 232: 82-88.
[67] TODD A D, BIELAWSKI C W. Thermally reduced graphite oxide reinforced polyethylene composites: A mild synthetic approach[J]. Polymer, 2013, 54(17): 4427-4430.
[68] STURZEL M, KEMPE F, THOMMAN Y, et al. Novel graphene UHMWPE nanocomposites prepared by polymerization filling using single-site catalysts supported on functionalized graphene nanosheet dispersions[J]. Macromolecules, 2012, 45 (17): 6878-6887.
[69] CHOI B, LEE J, LEE S J, et al. Generation of ultra-high-molecular-weight polyethylene from metallocenes immobilized onto N-Doped graphene nanoplatelets[J]. Macromolecules, 2013, 34 (6): 533-538.
[70] ALEXANDRE M, MARTIN E, DUBOIS P, et al. Use of metallocenes in the polymerization-filling technique with production of polyolefin-based composites[J]. Macromolecular Rapid Communications, 2000, 21(13): 931-936.
[71] ALEXANDRE M, MARTIN E, DUBOIS P, et al. Polymerization-filling technique: An efficient way to improve the mechanical properties of polyethylene composites[J]. Chemistry of Materials, 2001, 13(2): 236-237.
[72] HE F A, ZHANG L M. New polyethylene nanocomposites prepared by in-situ polymerization method using nickel a-diimine catalyst supported on organo-modified ZnAl layered double hydroxide[J]. Composites Science and Technology, 2007, 67(15-16): 3226-3232.
[73] HE F A, ZHANG L M. Organo-modified ZnAl layered double hydroxide as new catalyst support for the ethylene polymerization[J]. Journal of Colloid and Interface Science, 2007, 315(2): 439-444.
[74] MOBTEIL V, STUMBAUM J, THOMANN R, et al. Silica/polyethylene nanocomposite particles from catalytic emulsion polymerization[J]. Macromolecules. 2006, 39 (6): 2056-2062.
[75] WANG X, WU Q Y, DONG J Y, et al. Characterization of polyethylene/kaolin composites by polymerization filling with Cp2ZrCl2/MAO catalyst system[J]. Journal of Applied Polymer Science, 2002, 85(14): 2913-2921.
[76] ZHANG F, KARAKI T, ADACHI M, et al. Preparation of ferroelectric (Pb,Sr)TiO3/polyethylene nanocomposites and their dielectric properties[J]. Japanese Journal of Applied Physics Part 1-Regular Papers Brief Communications & Review Papers. 2006, 45 (3A): 1873-1876.
[77] JONGSOMJIT B, PANPRANOT J, PRASERTHDAM P. Effect of nanoscale SiO2and ZrO2as the fillers on the microstructure of LLDPE nanocomposites synthesized via in situ polymerization with zirconocene[J]. Materials Letters. 2007, 61 (6): 1376-1379.
[78] CHAICHANA E, JONGSOMJIT B, PRASERTHDAM P. Effect of nano-SiO2particle size on the formation of LLDPE/SiO2nanocomposite synthesized via the in situ polymerization with metallocene catalyst[J]. Chemical Engineering Science, 2007, 62 (3): 899-905.
[79] JONGSOMJIT B, PANPRANOT J, PRASERTHDAM P. LLDPE/nano-silica composites synthesized via in situ polymerization of ethylene/1-hexene with MAO/metallocene catalyst[J]. Journal of Materials Science, 2005, 40(8): 2043-2045.
[80] JONGSOMJIT B, PANPRANOT J, OKADA M, et al. Characteristics of LLDPE/ZrO2nanocomposite synthesized by in-situ polymerization using a zirconocene/MAO catalyst[J]. Iranian Polymer Journal, 2006, 15(5): 433-439.
[81] DESHARUN C, JONGSOMJIT B, PPRASERTHDAM P, et al. Study of LLDPE/alumina nanocomposites synthesized by in situ polymerization with zirconocene/d-MMAO catalyst[J]. Catalysis Communications. 2008, 9(4): 522-528.
[82] CHAICHANA E, NGOWTHANAWAT P, MEKASUWANDUMRONG O, et al. Catalytic performance of ZnO nanoparticle in formation of LLDPE/ZnO nanocomposites[J]. Iranian Polymer Journal, 2012, 21(1): 51-63.
[83] CHAICHANA E, PATHOMSAP S, MEKASUWANDUMRONG O, et al. LLDPE/TiO2nanocomposites produced from different crystallite sizes of TiO2via in situ polymerization[J]. Chinese Science Bulletin, 2012, 57(17): 2177-2184.
[84] CHENG W X, MIAO W, PENG J, et al. Synthesis of silica/polyolefin nanocomposites via two-step method[J]. Iranian Polymer Journal, 2009, 18(5): 365-371.
[85] COVARRUBIASA C, QUIJADAA R. Preparation of aluminophosphate/polyethylene nanocomposite membranes and their gas permeation properties[J]. Journal of Membrane Science. 2010, 358(1-2): 33-42.
[86] WANG N, SHI Z X, PENG J, et al. The influence of modification of mesoporous silica with polyethylene via in situ Ziegler-Natta polymerization on PE/MCM-41 nanocomposite[J]. Journal of Composite Materials, 2008, 42(12): 1151-1157.
[87] KALEEL S H A, BAHULEYAN B K, MASIHULLAH J, et al. Thermal and mechanical properties of polyethylene/doped-TiO2nanocomposites synthesized using in situ polymerization[J]. Journal of Nanomaterials, 2011: 964353.
[88] WANG L, FENG L X, XIE T. Novel magnetic polyethylene nanocomposites produced by supported nanometre magnetic Ziegler-Natta catalyst[J]. Polymer International, 2000, 49(2): 184-188
[89] WANG L, YUAN Y L, FENG L X. Preparation of novel magnetic polyethylene through polymerization in situ using CrxFe3-xO4/AlR3/TiCl4supported nanometer magnetic Ziegler-Natta catalyst[J]. Polymer Journal, 1999, 49(12): 1281-1283.
[90] WANG L, FENG L X, YANF S L. Studies on the preparation of new magnetic polyolefins using nanometer magnetic Ziegler-Natta catalyst[J]. Journal of Applied Polymer Science. 1999, 71(12): 2807-2090.
[91] 江洪流, 魏珊珊, 胡揚劍, 等. 負載型磁性納米二亞胺鎳催化劑制備新型磁性支化聚乙烯[J]. 高分子材料科學與工程. 2007, 23(2): 246-249.(JIANG Hongliu, WEI Shanshan, HU Yangjian, et al. Preparation of branched magnetic polyethylenes using nickelα-diimine complex covalently supported on nanometer magnetic support[J].Polymer Materials Science and Engeering, 2007,23(2):246-249.)
[92] PARK H J, KWAK S Y, KWAK S, et al. Wear-resistant ultra high molecular weight polyethylene/zirconia composites prepared by in situ Ziegler-Natta polymerization[J]. Macromolecular Chemistry and Physics. 2005, 18(9): 945-950.
[93] POTSCHKE P, BHATTAXHARYYA A R, JANKE A. Carbon nanotube-filled polycarbonate composites produced by melt mixing and their use blends with polyethylene[J]. Carbon, 2004,42(5-6):965-969.
A Review for Preparation of High Performance Polyethylene-Based Composites by In-situ Polymerization
HE Fuan1, 2, ZHANG Liming1
(1.InstituteofPolymerScience,CollegeofChemistryandChemicalEngineering,SunYat-senUniversity,Guangzhou510275,China;2.CollegeofChemicalEngineering,GuangdongUniversityofPetrochemicalTechnology,Maoming525000,China.)
In this review, recent progress on the preparation of high performance polyethylene-based composites by in-situ polymerization was commented, involving mainly clay/polyethylene composites, carbon nanotubes/polyethylene composites and graphite-based fillers/polyethylene composites. In particular, some routes to the in-situ polymerization in the presence of supported catalysts were summarized. The morphologies, structure and properties of resultant polyethylene-based composites were also discussed. In addition, some challenges for the development of high performance polyethylene-based composites were pointed out.
polyethylene; composites; in-situ polymerization; modifying agents
2014-10-20
廣東省自然科學基金研究團隊項目(039184)和教育部新世紀優秀人才支持計劃項目(NCET-04-0810)資助 第一作者: 何富安,男,研究員,博士,從事高分子材料設計制備與性能研究
張黎明,男,教授,博士,從事高分子材料設計制備與性能研究;Tel:020-84112354;E-mail:ceszhlm@mail.sysu.edu.cn
1001-8719(2015)02-0369-21
O63
A
10.3969/j.issn.1001-8719.2015.02.017
猜你喜歡
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
纖維復合材料(2018年3期)2018-04-25 07:22:58
電子測試(2017年11期)2017-12-15 08:57:13
中國塑料(2016年12期)2016-06-15 20:30:07
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
中國塑料(2015年11期)2015-10-14 01:14:14
中國塑料(2015年9期)2015-10-14 01:12:17
中國塑料(2015年4期)2015-10-14 01:09:19
