腺苷鈷胺有關物質分析方法的改進及雜質研究
2015-07-07 14:57:44楊洪淼廖海明任雪范慧紅
中國生化藥物雜志 2015年6期
楊洪淼,廖海明,任雪,范慧紅
(中國食品藥品檢定研究院,北京 100050)
?
腺苷鈷胺有關物質分析方法的改進及雜質研究
楊洪淼,廖海明,任雪,范慧紅Δ
(中國食品藥品檢定研究院,北京 100050)
目的 改進腺苷鈷胺有關物質的分析方法,并對腺苷鈷胺光降解的特性進行研究。 方法 采用色譜條件為Merck Hibar C18色譜柱(4.6 mm×250 mm,5 μm),0.05 mol/L磷酸二氫鉀溶液:乙腈(90:10)-80%乙腈為流動相進行梯度洗脫,檢測波長為260 nm的高效液相色譜法測定經光照后的腺苷鈷胺固體和水溶液2種不同狀態的有關物質,比較其光穩定性;采用LC-MS方法測定其主要的3種光降解雜質。結果 梯度洗脫條件可以使腺苷鈷胺的光降解雜質更好的分離開,精密度和重復性實驗結果由RSD=3.8%(n=6)和RSD=38%(n=5)提高至RSD=0.2%(n=6)和RSD=8%(n=5)。此外腺苷鈷胺溶液對光非常敏感,配制時應注意嚴格避光操作。2種光降解雜質為腺苷和羥鈷胺,相對響應因子分別為2.5和0.7。結論 改進后的方法專屬性、耐用性和重復性均很好,可以更好的為腺苷鈷胺的質量控制服務。
腺苷鈷胺;高效液相色譜;有關物質;液相色譜-質譜
腺苷鈷胺化學名為5,6-二甲基苯并咪唑基-5’-脫氧腺嘌呤核苷基鈷胺,分子式為C72H100CoN18O17P,分子量1579.60,CAS號13870-90-1。腺苷鈷胺及其片劑收載于《中國藥典》2010年版二部,其片劑同時收載于《國家基本藥物目錄》(2012版)。氰鈷胺、羥鈷胺、腺苷鈷胺及甲鈷胺均屬于維生素B12類物質,但只有腺苷鈷胺和甲鈷胺是活性輔酶形式,在人體內可直接發揮生物活性。臨床上廣泛應用于神經炎、神經麻痹等各種神經損傷的修復[1-4]。
本文將《中國藥典》2010年版中腺苷鈷胺項下有關物質分析方法加以改進,可以將腺苷鈷胺的光降解雜質更好的分離開,從而有效提高雜質定量的準確性和重復性。由于腺苷鈷胺具有較強的光不穩定特性,本文考察了固體和水溶液2種不同狀態的光穩定性,為在檢驗過程中應如何進行避光操作提供了數據基礎。并對主要的3種光降解雜質進行了LC-MS測定,最終確定了2種雜質的結構,進一步測定了這2種已知雜質的相對響應因子。
1 材料與方法
1.1 藥品與試劑 腺苷鈷胺(批號140658-201302,含量98.5%)、腺苷(批號110879-200202,含量99.0%)(購自中國食品藥品檢定研究院)。羥鈷胺素(貨號H7126,含量90.3%)、5’-脫氧腺苷(貨號D1771,含量≥90.0%)(購自Sigma Alrich公司)。乙腈(購自Fisher試劑公司,色譜純),磷酸、磷酸二氫鉀(購自國藥集團化學試劑有限公司,分析純)。
1.2 儀器 Agilent 1100 液相色譜儀(包括G1311A四元泵、G1329自動進樣器、G1316A柱溫箱、G1314A VWD檢測器、HP ChemStation for LC)(購自美國Agilent公司);MP230型pH計(購自瑞士Mettler Toledo公司);LTQ orbitrap型高分辨率液質聯用儀(購自美國Thermo公司)。
1.3 腺苷鈷胺有關物質測定方法的改進
1.3.1 色譜條件及溶液的配制:
① 色譜條件:色譜柱:Merck Hibar C18柱(4.6 mm×250 mm,5 μm);流動相:流動相A為0.05 mol/L磷酸二氫鉀溶液(用磷酸調節pH值至3.2):乙腈(90:10),流動相B為乙腈:水(80:20);檢測波長:260 nm;柱溫:35 ℃;進樣量10 μL;流速:1 mL/min。
② 供試品溶液配制:稱取本品約25 mg,置25 mL量瓶中,加水適量,超聲處理使溶解并加水稀釋至刻度。避光操作。
③ 對照溶液配制:精密量取供試品溶液1 mL,至100 mL量瓶中,加水稀釋至刻度。采用自身對照法進行定量。
1.3.2 梯度的改進:藥典方法為采用0.05 mol/L磷酸二氫鉀溶液(用磷酸調節pH值至3.2):乙腈(85:15),進行恒梯度洗脫;改進后的色譜條件為梯度洗脫,條件為B相:0~7 min,2%→9%;7~25 min,9%;25~25.1 min,9%→2%;25~35 min,2%。
1.3.3 精密度試驗:將同一份供試品溶液,分別采用藥典方法及改良方法,各連續進樣6針,計算主峰前雜質峰面積總和的RSD。
1.3.4 重復性試驗:配制5份濃度約為1 mg/mL的供試品溶液。分別采用采用藥典方法及改良方法進樣,采用峰面積歸一化法計算主峰前面雜質的含量總和。
1.3.5 方法的耐用性考察:
① 色譜柱的影響:除Merck Hibar C18柱外,還考察了Venusil XBP C18柱(4.6 mm×250 mm,5 μm)和華譜Unitary C18柱(4.6 mm×250 mm,5 μm)。
② 流動相pH的影響:將流動相A中的磷酸鹽緩沖液的pH調至4.5,其他條件不變進行實驗。
1.4 腺苷鈷胺穩定性的研究
1.4.1 光照破壞試驗:
① 固體的光不穩定性試驗:稱取腺苷鈷胺約50 mg,置稱量瓶中,室內(非陽光直射、非日光燈照射,光度約600 xL)放置0.5 h后,進行有關物質測定。同時測定了60 ℃減壓干燥至恒重后的樣品(使用透明稱量瓶,將真空干燥箱的可視窗口用白紙蓋住,同時將干燥器置于暗處冷卻,但其他操作步驟非避光操作)的有關物質。
② 溶液的光不穩定性試驗:將1 mg/mL的腺苷鈷胺溶液放置于敞口燒杯中,室內(光度條件同上)放置0.5 h后,按改良后的方法進行有關物質測定。
1.4.2 酸堿、氧化、高溫破壞試驗:
① 酸破壞試驗溶液配制:稱取本品約5 mg,加入1 mol/L鹽酸溶液1.0 mL,避光放置30 min,加1 mol/L氫氧化鈉溶液1.0 mL中和,再加水稀釋至5 mL。
② 堿破壞試驗溶液配制:稱取本品約5 mg,加入2 mol/L氫氧化鈉溶液1.0 mL,避光放置45 min,加1 mol/L鹽酸溶液2.0 mL中和,再加水稀釋至5 mL。
③ 高溫破壞試驗溶液配制:稱取本品約5 mg,加水5.0 mL溶解,置于90 ℃烘箱中加熱15 min。
④ 氧化破壞試驗溶液配制:稱取本品約5 mg,加水2 mL溶解,加0.1%氯胺T溶液0.4 mL,再加0.05 mol/L鹽酸溶液0.1 mL,放置1 min,再加水至5 mL。
⑤ 分別取上述破壞試驗溶液20 μL,按改良色譜條件測定。
1.5 腺苷鈷胺的雜質研究
1.5.1 LC-MS法對腺苷鈷胺光降解雜質的鑒定:
① 色譜條件:色譜柱為華譜Unitary C18柱(2.1 mm×10 mm,2.8 μm);流動相A為0.1%醋酸-乙腈溶液(90:10),流動相B為80%乙腈溶液,梯度洗脫(0~15 min,B相0%→15%);流速:0.2 mL/mL;檢測波長:260 nm;柱溫:35 ℃;進樣5 μL。
② 質譜條件:正離子檢測模式;數據采集范圍m/z=100~2000;毛細管電壓:4 kV;干燥器流速:8 mL/min;干燥器溫度:350 ℃;霧化氣壓力:0.24 MPa;裂解電壓:110 V;碎片碰撞能量:20 eV。
③ 樣品溶液配制:取腺苷鈷胺加水制成1 mg/mL的樣品溶液。
1.5.2 雜質腺苷及羥鈷胺的進一步確證:取腺苷對照品及羥鈷胺對照品與腺苷鈷胺在相同的色譜條件下進行液相測定。
1.5.3 已知雜質響應因子的測定: 精密稱取腺苷鈷胺、腺苷、羥鈷胺各10 mg,置于同一10 mL量瓶中,加水制得儲備液。精密量取儲備液適量,加水稀釋分別制成濃度為0.6125、1.25、2.5、5、10、20、40 μg/mL的系列溶液,分別精密量取上述溶液10 μL注入液相色譜儀,記錄色譜圖。以濃度為橫坐標(C),以峰面積為縱坐標(A),進行線性回歸。
2 結果
2.1 梯度的洗脫 采用藥典方法測定腺苷鈷胺的有關物質時發現,供試品溶液的色譜圖在死體積后會出現一簇色譜峰,見圖1A。進一步研究發現這些色譜峰并非溶劑峰,而是腺苷鈷胺光降解的主要雜質峰。并且這些峰的峰面積在同份供試品溶液的不同次進樣之間或是相同濃度的不同份供試品溶液之間,積分得到的結果變異性均較大,導致有關物質測定結果的重復性差。
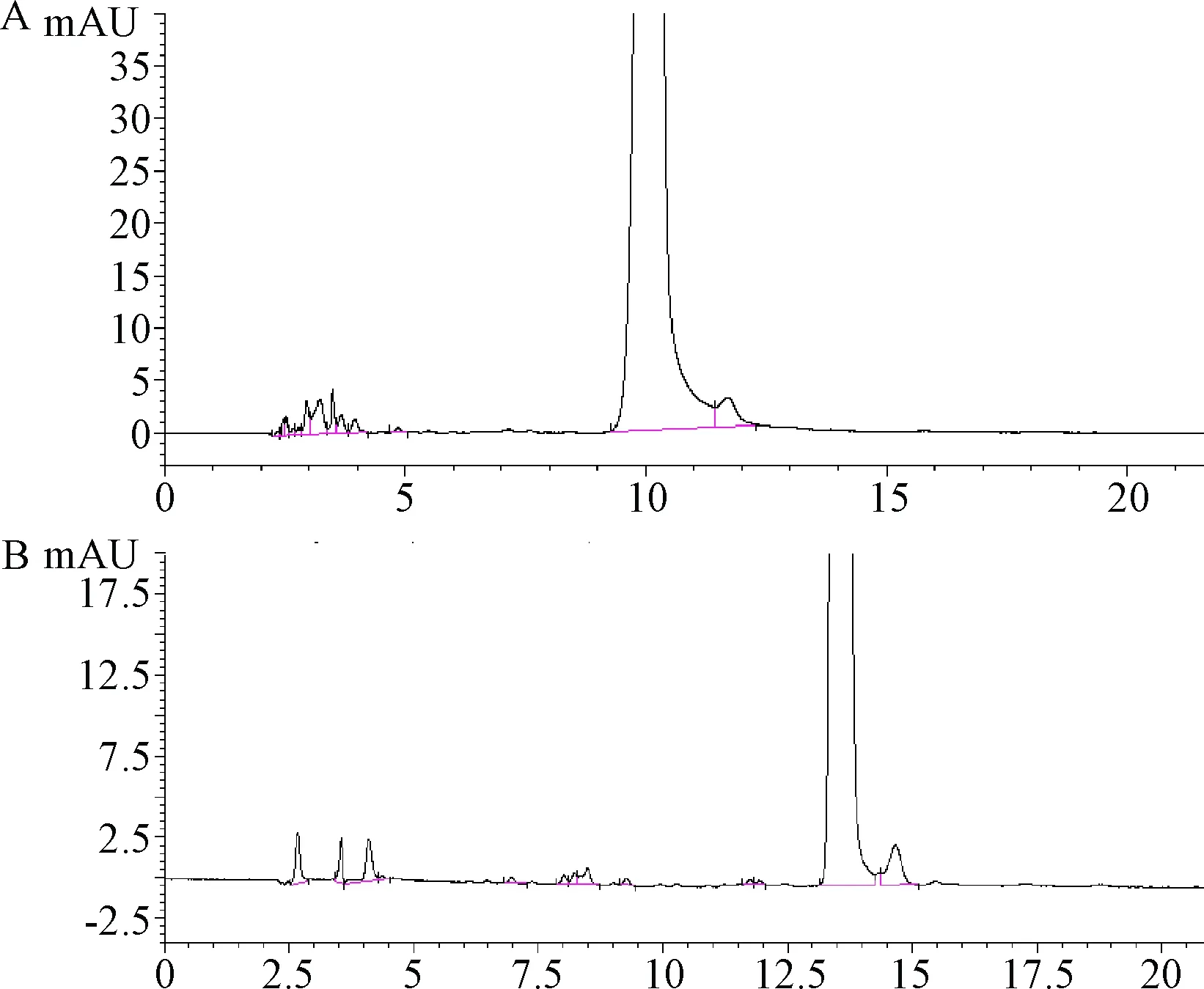
圖1 2種方法進行有關物質分析的色譜圖A.藥典方法;B.新方法Fig.1 Chromatogram of related substances analysis using two methodsA.Pharmacopeia method;B.New method
嘗試將恒梯度洗脫改為梯度洗脫從而將前面的雜質峰分離,梯度洗脫條件所得色譜圖,見圖1B。前端可清楚地分出3個主要雜質峰,分離度良好。
2.2 精密度試驗和重復性試驗 精密度試驗和重復性試驗結果均表明新方法對結果有顯著的改善,見表1。

表1 2種方法進行的精密度試驗與重復性試驗的結果Tab.1 Results of precision test and repeatability test in two methods
2.3 方法的耐用性試驗
2.3.1 色譜柱的影響:不同色譜柱所得到色譜圖中雜質的分離度均良好,見圖2。
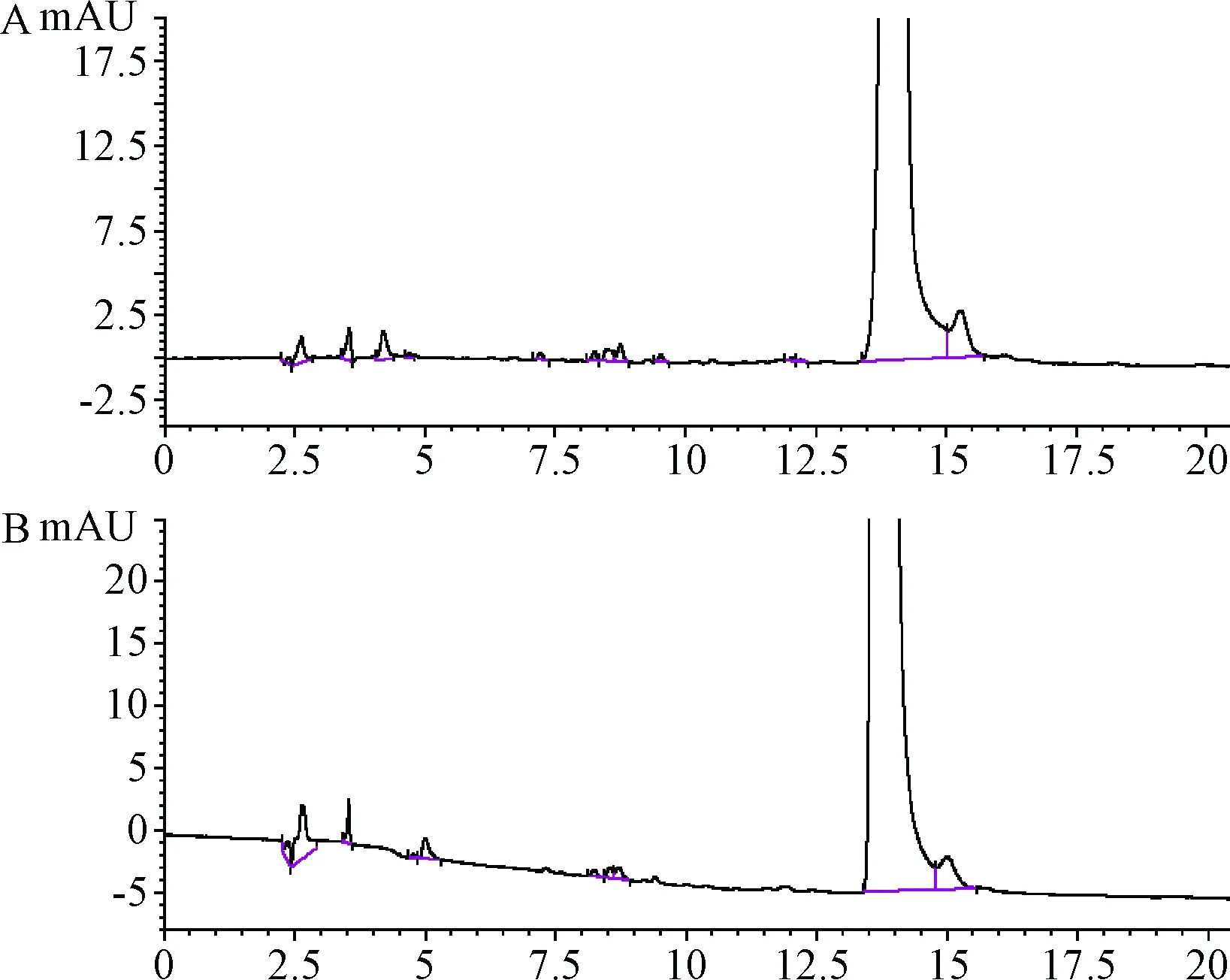
圖2 不同色譜柱進行有關物質分析的色譜圖A.Venusil XBP C18柱;B.華譜Unitary C18柱Fig.2 Chromatogram of related substances analysis by different columnsA.Venusil XBP C18 column;B.Huapu Unitary C18 column
2.3.2 流動相pH的影響:所得色譜圖中各峰的峰形稍差,但色譜圖前端的雜質依然可以分開,見圖3。
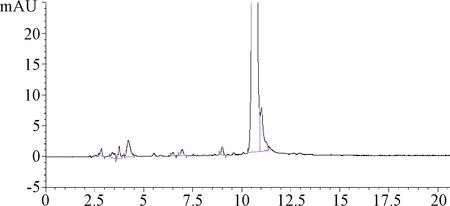
圖3 流動相A的pH為4.5時進行有關物質分析的色譜圖Fig.3 Chromatogram of related substances analysis with pH4.5 of mobile phase A
2.4 光照破壞試驗
2.4.1 固體的光破壞條件:經過光照后的腺苷鈷胺固體及60 ℃減壓干燥至恒重后的樣品雜質均未見明顯增加。
2.4.2 溶液的光不穩定性試驗:腺苷鈷胺溶液經光照后,色譜圖前端3個雜質峰面積明顯增加,腺苷鈷胺的峰面積降低約10%,見圖4。而將腺苷鈷胺溶液放置于黑暗的環境中(如液相色譜儀的可遮光的進樣盤內),放置12 h后,雜質也未見明顯增加。
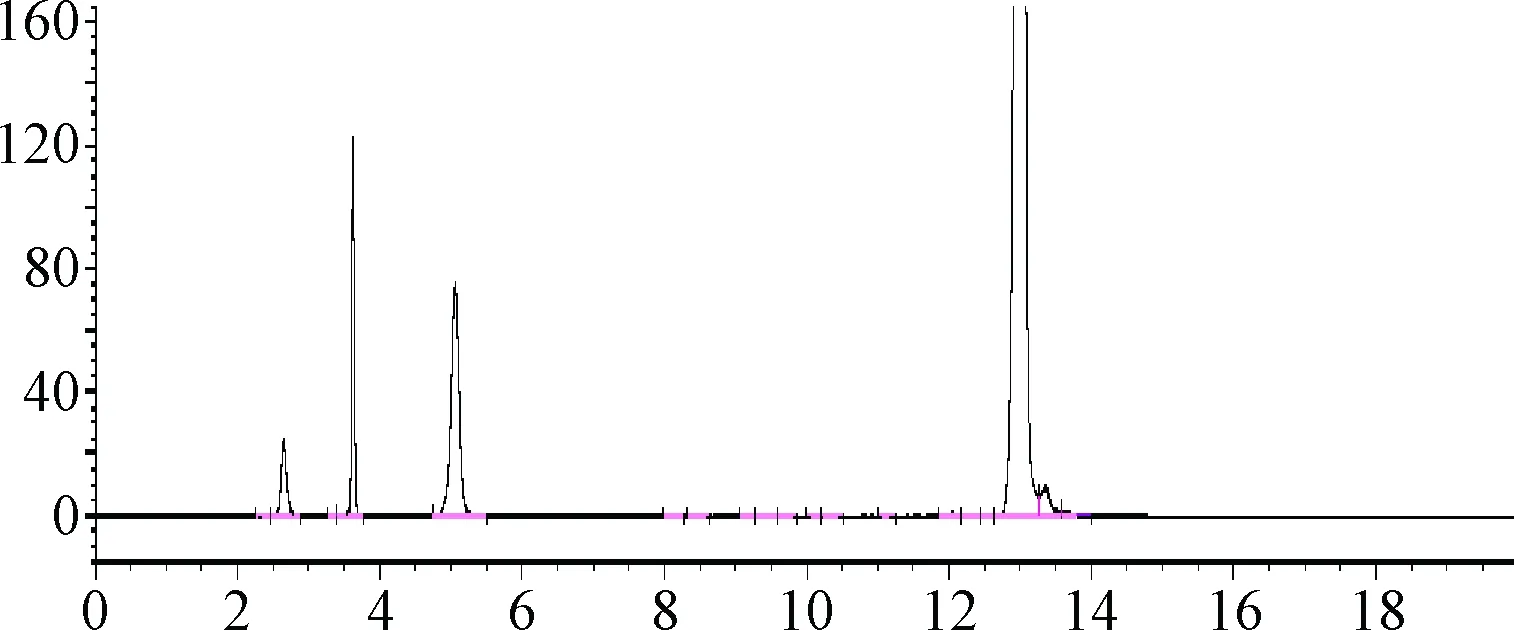
圖4 腺苷鈷胺溶液光降解試驗色譜圖Fig.4 Chromatogram of light degradation test of cobamamide solution
2.5 酸堿、氧化、高溫破壞試驗 結果表明本品除對光不穩定,對酸、堿、加熱和氧化也不穩定,而本方法可以將降解雜質與主成分進行較好的分離,尤其可以將色譜圖前端產生的主要降解雜質之間進行較好分離,更利于雜質的控制。氧化破壞試驗條件是參照《中國藥典》維生素B12品種[4]項下有關物質檢查中系統適用性溶液的配制方法,所得色譜圖與維生素B12類似[5-6],主峰后出現降解雜質峰,且分離度良好。本實驗結果說明改良后的有關物質測定方法專屬性良好。見圖5。
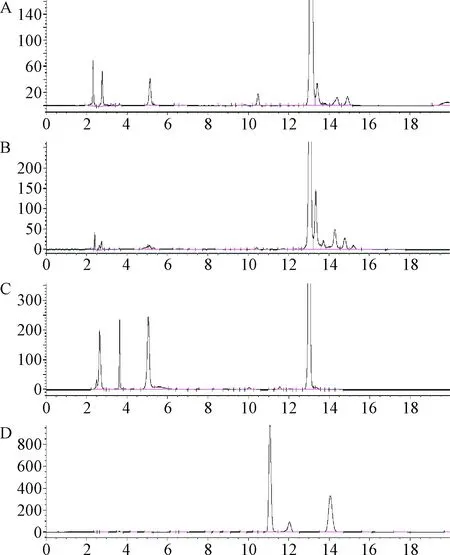
圖5 腺苷鈷胺酸、堿、高溫和氧化降解試驗色譜圖A.酸降解試驗;B.堿降解試驗;C.高溫降解試驗;D.氧化降解試驗Fig.5 Chromatogram of acid, alkali, high temperature and oxidative degradation test of cobamamideA.Acid degradation test;B.Alkaline degradation test;C.Heat degradation test;D.Oxidative degradation test
2.6 質譜鑒定 紫外光譜圖和總離子流圖見圖6。腺苷鈷胺光降解雜質所得的質譜圖見圖7。雜質峰1準分子離子峰為284.10,比腺苷分子量增加16,將此碎片進行LC-MS-MS測定,可得準分子離子峰136.13,與腺嘌呤[M+H]+的分子量相同。初步推測雜質峰1的結構可能為在腺苷的分子結構中核糖的上面加合了一個氧原子,但具體的位置還需要更多的試驗證明;雜質峰2樣品溶液中隨雜質2濃度不同時,會測得不同的準分子離子峰284.10或250.22,推測為該雜質在離子源內發生反應,最常見的源內反應為加氧或脫水的反應,由于腺苷的分子量為267.24,所以不同的準分子離子峰可能為腺苷的[M+O+H]+峰或[M-H2O+H]+峰,由此推測出雜質峰2可能為腺苷;雜質峰3所得的準分子離子峰為664.78,由羥鈷胺的分子量1346.32得出該準分子離子峰可能為羥鈷胺的[M-H2O+2H]/2+峰,雜質峰3推測為羥鈷胺。
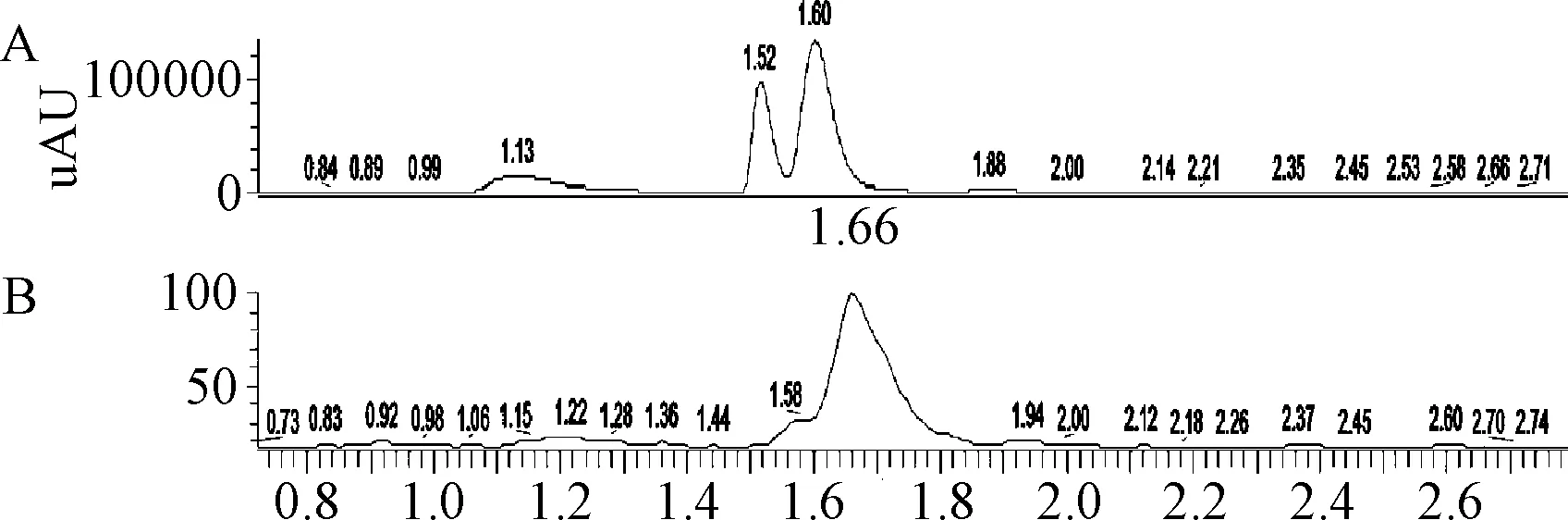
圖6 腺苷鈷胺光降解雜質LC-MS測定圖A.紫外光譜圖;B.總離子流圖Fig.6 LC-MS chromatogram of light degradation purities of cobamamideA.UV chromatogram;B.TIC chromatogram
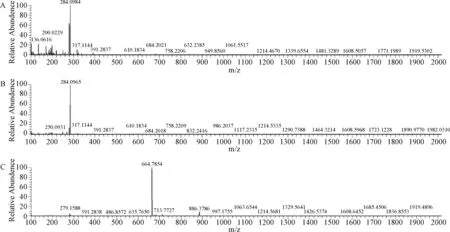
圖7 腺苷鈷胺光降解雜質所得的質譜圖A.雜質峰1;B.雜質峰2;C.雜質峰3Fig.7 MS spectra for light degradation purities of cobamamideA.Impurity peak 1;B.Impurity peak 2;C.Impurity peak 3
2.7 雜質腺苷及羥鈷胺的進一步確證 腺苷與雜質2的出峰時間一致,并使用DAD檢測器得到峰的紫外吸收光譜,2者也是重合的,而羥鈷胺與雜質3的出峰時間一致并且峰的紫外吸收光譜重合,見圖8。由此進一步確證了腺苷鈷胺的光降解產物中含有腺苷及羥鈷胺。
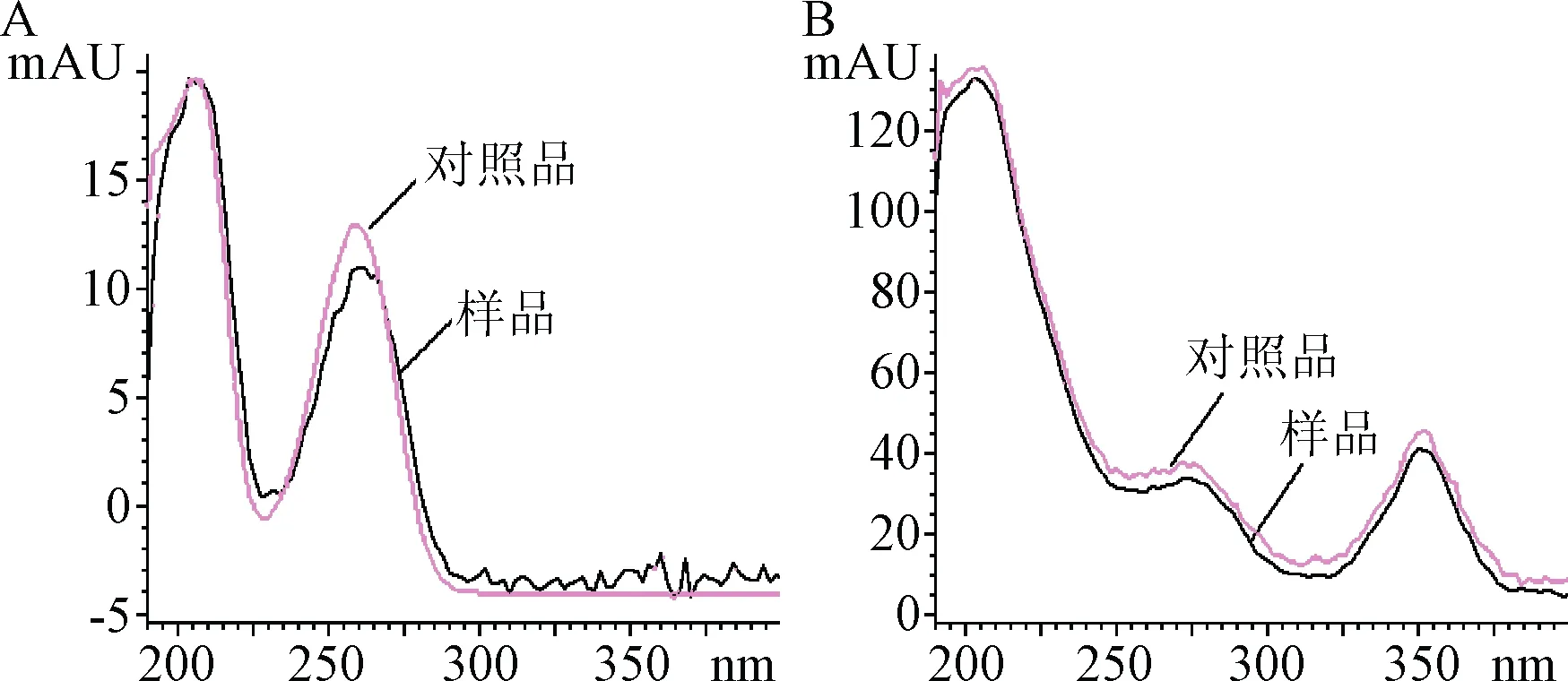
圖8 對照品與樣品紫外吸收光譜比較圖A.腺苷;B.羥鈷胺Fig.8 UV absorption spectra of the reference standard and sampleA.Adenosine;B.Hydroxycobalamin
2.8 已知雜質響應因子的測定 腺苷鈷胺、腺苷和羥鈷胺線性方程(n=7)分別為:A=11.40C (r=0.997);A=28.66C+5.590 (r=0.999);A=7.505C+2.969(r=0.999)。經計算,相對于主成分腺苷鈷胺,腺苷的響應因子為2.5;羥鈷胺的響應因子為0.7。
3 討論
根據查閱的腺苷鈷胺有關物質分析的文獻[7-10],本文首次采用梯度洗脫的方法進行分析。改進后的方法重復性、專屬性及耐用性均良好,可以更加準確的對其雜質進行定量,從而達到更嚴格的藥品質量控制。此外將腺苷鈷胺的光降解雜質在色譜中分開,也為進一步雜質研究提供了基礎。
通過對腺苷鈷胺固體和水溶液光不穩定性的比較可得,水溶液對光極為敏感,而固體相對穩定。所以對腺苷鈷胺固體進行稱量時可以在普通天平室操作,但進行溶液配制時需要在暗室內操作,尤其對配制有關物質測定的供試品溶液時,此點尤為重要。
腺苷鈷胺光降解雜質方面的研究較少。曾有文獻報道5’-脫氧腺苷和羥鈷胺可能是腺苷鈷胺的光降解雜質[11],但通過高效液相色譜出峰時間的比較得出5’-脫氧腺苷并不是本文通過實驗證明了腺苷和羥鈷胺是腺苷鈷胺的主要光降解產物,而已知雜質相對響應因子的測定可以進一步提高有關物質定量的準確性。
[1] 劉新通.腺苷鈷胺的臨床應用[J].中國處方藥,2004,26(5):20-21.
[2] 王志良,冷健,崔紅燕.腺苷鈷胺的臨床應用進展[J].中國藥事,2006,20(2):122-125.
[3] 尚學軍,胡衛國,姚福鑫,等.腺苷鈷胺的藥理及臨床作用[J]臨床合理用藥,6(8):80.
[4] 中國藥典2010年版,ISBN 978-7-5067-4483-6[S].北京:中國醫藥科技出版社,2010:1.
[5] 劉毅,岳志華.HPLC測定維生素B12注射液的含量和有關物質[J].中國藥學雜志,2010,25(4):304-306.
[6] 劉毅,岳志華.維生素B12有關物質檢查方法的探討[J].藥物分析雜志,2010,30(3):550-552.
[7] 殷莉莉,廉英,郭妍,等.高效液相色譜法測定腺苷鈷胺片中的羥鈷胺素含量[J].中南藥學,2014,12(7):677-679.
[8] 晏敏紅,麻軍法.注射用腺苷鈷胺有關物質測定方法改進[J].海峽藥學,2013,25(8):62-65.
[9] 王志良,王芳,尹書芳,等.HPLC法測定注射用腺苷鈷胺有關物質[J].中國藥事,2006,20(8):492-493.
[10] 陳橋.HPLC測定腺苷鈷胺有關物質[J].河北化工,2009,32(6):62-63.
[11] 封淑華,李力更,王麗萍.腺苷鈷胺的光解研究[J].中國藥科大學學報,1997,28(1):183-186.
(編校:王冬梅)
Improved analysis of related substances of cobamamide and the impurities research
YANG Hong-miao, LIAO Hai-ming, REN Xue, FAN Hui-hongΔ
(National Institutes for Food and Drug Control, Beijing 100050,China)
ObjectiveTo improve the related substances analysis method of cobamamide.And to further research light degradation characteristics of cobamamide.MethodsDetermined the related substances of solid and aqueous solution of cobamamide after light degradation by high performance liquid chromatography(HPLC) conditions,which is Merck Hibar C18column (4.6 mm× 250 mm, 5 μm), 0.05 mol/L KH2PO4solution:acetonitrile (90:10)-80% acetonitrile as mobile by gradient elution and detection wavelength 260 nm.And compared their light stability.The main three kinds of light degradation impurities were determinate from LC-MS.ResultsGradient elution made the light degradation impurities separate better.The results of precision and reproducibility tests increased to RSD=0.2% (n=6) and RSD=8% (n=5) from RSD=3.8% (n=6) and RSD=38%(n=5).Cobamamide solution was very sensitive to light, the preparation should be strict dark operation.Two of the light degradation impurities were adenosine and hydroxycobalamin, with the relative response factor 2.5 and 0.7.ConclusionNew method is specific, durable and reproducible, which can be used for quality control of cobamamide.
cobamamide; HPLC; related substances; LC-MS
楊洪淼,女,碩士,助理研究員,研究方向:生化藥品的質量分析和質量標準的研究,E-mail:yanghongmiao@163.com;范慧紅,通訊作者,女,博士,研究員,研究方向:生化藥品的質量控制和質量標準的研究,E-mail:shenghuayaoshi@126.com。
R917
A
1005-1678(2015)06-0157-05
