2,3-二取代-5-乙烯基-4,5-二氫呋喃的合成
2015-07-19 12:35:43肖利周志鵬王海飛胡舜欽
湖南工業大學學報 2015年1期
肖利,周志鵬,王海飛,胡舜欽
(湖南工業大學包裝與材料工程學院,湖南株洲412007)
2,3-二取代-5-乙烯基-4,5-二氫呋喃的合成
肖利,周志鵬,王海飛,胡舜欽
(湖南工業大學包裝與材料工程學院,湖南株洲412007)
為解決合成呋喃環需加入較多較昂貴的金屬催化劑或者堿的問題,利用-酮酯與對稱的烯丙基二碳酸酯為原料,以烯丙基二碳酸酯的物質的量為用量標準,在以占標準用量2%的四(三苯基膦)鈀與占標準用量2%的1,2-雙- (二苯基膦)乙烷的作用下,合成了一系列的2,3-二取代-5-乙烯基-4,5-二氫呋喃衍生物。試驗結果表明,取代基為吸電子基團時反應具有較高的產率,通過改變取代基,已得到了9個不同取代的2,3-二取代-5-乙烯基-4,5-二氫呋喃衍生物,所有產物結構均經1H及13C NMR確證。
-酮酯;烯丙基二碳酸酯;二氫呋喃
0 引言
呋喃(furan)是一種含有4個碳原子和1個氧原子的五元雜環有機物,其結構單元廣泛存在于天然產品[1-2]、藥品[3]、香料[4]和農用產品[5]等中,此結構單元的存在使得這些物質具有呋喃的結構和功能特性[6],如抗癌、抗艾滋病和抗濾過性病原體等。
在呋喃雜環的制備方法[7-13]中,金屬試劑催化合成呋喃環衍生物早已引起人們的注意,選用的金屬一般是過渡金屬,如銅、金、銀和鈀等。由于金屬催化劑催化合成呋喃環衍生物具有催化劑用量少、反應條件溫和及操作簡便等優點,促使更多的合成化學家致力于研究發展更多直接有效的合成方法。1994年,Dixneuf小組[12]報道了利用Ru(PPh3) (p-cymene)Cl2催化底物烯炔醇合成呋喃環衍生物的方法,但此方法僅限于含有末端炔基的反應;2004年,Nishizawa小組[13]報道了利用Hg(OTF)2催化合成呋喃環衍生物的研究,此反應雖然反應速度較快,但是體系毒性較大;2007年,Tanimori小組[7]報道了利用[Pd (3-C3H5)Cl]2催化底物烯丙基二碳酸酯與底物-酮酯合成呋喃環的研究,此研究雖能進行反應,但生成物的產率都不高。
C-烯丙基化反應和O-烯丙基化反應是有機化合物分子中碳碳鍵和碳氧鍵形成反應中的比較重要的2個反應。本文擬根據烯丙基二碳酸酯與-酮酯通過分子間的C-烯丙基化反應與分子內的O-烯丙基化反應選擇性地制備呋喃環衍生物。雖然近年來已有部分文獻報道采用此方法[9]合成呋喃衍生物,但其采用較昂貴的催化劑或加入堿才能使反應進行,而本文僅以-酮酯與對稱的烯丙基二碳酸酯為原料,以烯丙基二碳酸酯的物質的量為用量標準,以占標準用量2%的四(三苯基膦)鈀與占標準用量2%的1,2-雙(二苯基膦)乙烷的作用下合成2,3-二取代-5-乙烯基-4,5-二氫呋喃,此反應具有催化劑用量少、反應條件溫和及操作簡便等優點。本文擬通過篩選不同配體與溶劑種類以及金屬催化劑與配體的用量為研究內容,研究它們對合成呋喃環衍生物的影響,確定最佳的反應條件和可能的反應機理,并通過1H及13C核磁共振(nuclear magnetic resonance,NMR)對合成的產物進行確證,以期獲得不同取代基的呋喃環衍生物。
1 試驗部分
1.1 試劑與儀器
石油醚、乙酸乙酯(ethyl acetate,AcOEt)、二氯甲烷(dichloromethane,DCM),均為分析純,天津津東天正精細化學試劑廠生產;甲苯(methylbenzene,Toluene)、四氫呋喃(terahydrofuran,THF)、1,2-二氯乙烷(1,2-dichloroethane,EDC)、甲醇(methyl alcohol,MeOH)、1,4-二氧六環、吡啶、碳酸二甲酯、氯甲酸乙酯、順式1,4-丁烯二醇,均為分析純,上海泰坦科技股份有限公司生產;氫化鈉,質量分數為60%,薩恩化學技術(上海)有限公司生產;1,2-雙(二苯基膦)乙烷(1,2-bis(diphenylphosphino)ethane,DPPE)、三苯基膦(triphenyl phosphine,PPh3)、1,1’-雙(二苯基膦)二茂鐵(1,1’-bis(diphenylphosphino)ferroce,DPPF)、1,4-雙(二苯基膦)丁烷(1,4-bis(diphenyphosphino)butane,DP P B)、1,3-雙(二苯基膦)丙烷(1,3-bis (diphenylphosphino) propane,DPPP)、四(三苯基膦)鈀((beta-4)-platinum,Pd(PPh3)4),均為分析純,薩恩化學技術(上海)有限公司生產;苯乙酮、對甲氧基苯乙酮、間甲氧基苯乙酮、2-萘乙酮、對氟苯乙酮、鄰氟苯乙酮、鄰溴苯乙酮、對溴苯乙酮、間溴苯乙酮,均為分析純,阿拉丁試劑(上海)有限公司生產。
核磁共振儀,Bruker AV-400 MHz型,瑞士Bruker公司生產;數顯智能控溫磁力攪拌器,SZCL-3B型,鞏義市予華儀器責任有限公司生產;分析電子天平,JA1003型,上海舜宇恒平科學儀器有限公司生產;三用紫外分析儀,ZF-6型,上海嘉鵬科技有限公司生產;遠紅外快速恒溫干燥箱,YHG-600S型,上海躍進醫療器械廠生產;旋轉蒸發儀,R201D型,鄭州市亞榮儀器有限公司生產。
1.2 試驗過程
本研究按照圖1所示的實驗流程圖制備呋喃環衍生物。首先,根據文獻合成-酮酯1a~1i與對稱的烯丙基二碳酸酯2,再將合成的-酮酯1a~1i與烯丙基二碳酸酯2在一定條件下反應合成相應的呋喃環衍生物3a~3i。
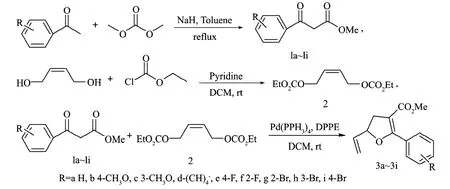
圖1 呋喃衍生物的合成流程圖Fig. 1The flowchart for furan derivatives synthesis
0℃條件下,在50 mL燒瓶中加入氫化鈉15 mmol與甲苯10 mL,攪拌10 min,再將含有相應取代基的苯乙酮5 mmol與甲苯2.5 mL的混合液逐滴加入燒瓶內,回流1 h;待溫度降至0℃后,將碳酸二甲酯10 mmol逐滴加入其中,滴加完全后回流3~6 h;采用薄層色譜法(thin-layer chromatography,TLC)點板跟蹤檢測至原料點消失,冷卻后滴加冰醋酸至糊狀物不再增加,再向其中加入冰水至糊狀物完全溶解。采用乙酸乙酯提取,并用無水硫酸鎂干燥,過濾,濃縮,柱層析純化得相應的-酮酯1a~1i。[14-16]
2)烯丙基二碳酸酯2的制備
在0℃條件下,將順式1,4-丁烯二醇10 mmol與吡啶2.5 mmol加入到含有33 mL二氯甲烷溶劑的燒瓶中,攪拌均勻,再將氯甲酸乙酯25 mmol逐滴加入其中;在0℃條件下攪拌10 min,再在室溫下攪拌1 h,采用薄層色譜法點板跟蹤檢測至原料點消失,加入二氯甲烷33 mL,用濃度為1 mol/L稀鹽酸33 mL進行3次萃取,萃取后的有機相用飽和碳酸鈉溶液33 mL進行3次洗滌,再用飽和氯化鈉溶液33 mL進行3次洗滌至中性;干燥,濃縮,柱層析純化得無色液體產物。[17-18]
3)呋喃環衍生物的合成
準確量取四(三苯基膦)鈀0.01 mmol、1,2-雙(二苯基膦)乙烷0.01 mmol與-酮酯1a~1(i0.75 mmol)加入到含有1.5 mL二氯甲烷溶劑的燒瓶中,攪拌均勻,再將0.5 mmol烯丙基二碳酸酯2加入燒瓶中,室溫攪拌4~6 h,采用薄層色譜法點板跟蹤檢測至原料點消失,反應體系不需其它操作,直接柱層析純化得油狀液體產物3a~3i。
1.3 產物的表征與檢測
合成的呋喃環衍生物經1H及13C NMR確認,其部分產物的核磁譜圖如圖2所示。
以2-苯基-3-羧基甲酯-5-乙烯基-4,5-二氫呋喃的核磁譜圖為例,從圖2a中13C NMR可看出,化學位移在100以下的歸屬于飽和基團上的碳,而化學位移在100以上的歸屬于不飽和基團上的碳,由13C NMR可以粗略地推算出碳原子的個數。再對圖2b中的1H NMR進行分析,可進一步確認,其化學位移在7.73~7.28之間的歸屬于苯環上的氫,化學位移在5.98~5.05之間的歸屬于烯基上的氫,化學位移在3.25~2.81之間的歸屬于亞甲基上的氫,化學位移為3.59的單峰歸屬于甲氧基上的氫。其他物質的譜圖,例如2-(4-甲氧基-苯基)-3-羧基甲酯-5-乙烯基-4,5-二氫呋喃(圖2c,2d)中的給電子取代基與2-間溴苯基-3-羧基甲酯-5-乙烯基-4,5-二氫呋喃(圖2e,2f)中的吸電子取代基,都可采用類似的方法進行確證。含有吸電子基團或給電子基團,其化學位移會有所變化,但不大,從氫譜和碳譜上看,含有給電子基團使部分氫和碳的化學位移向高場移動,而吸電子基團使其化學位移向低場移動。
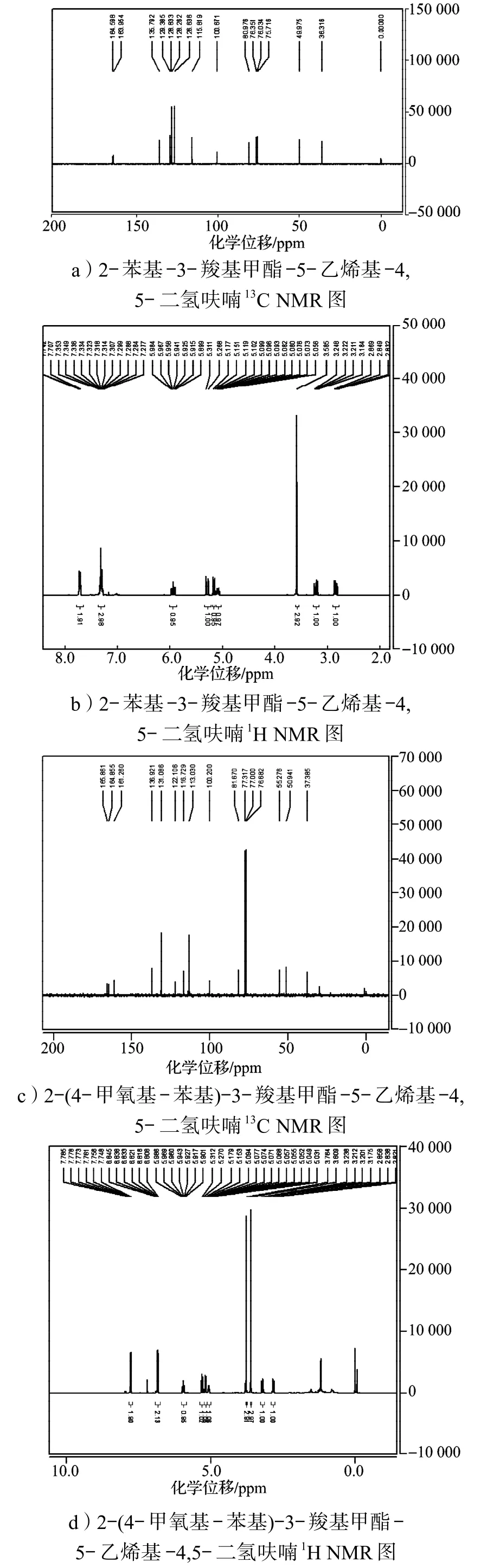
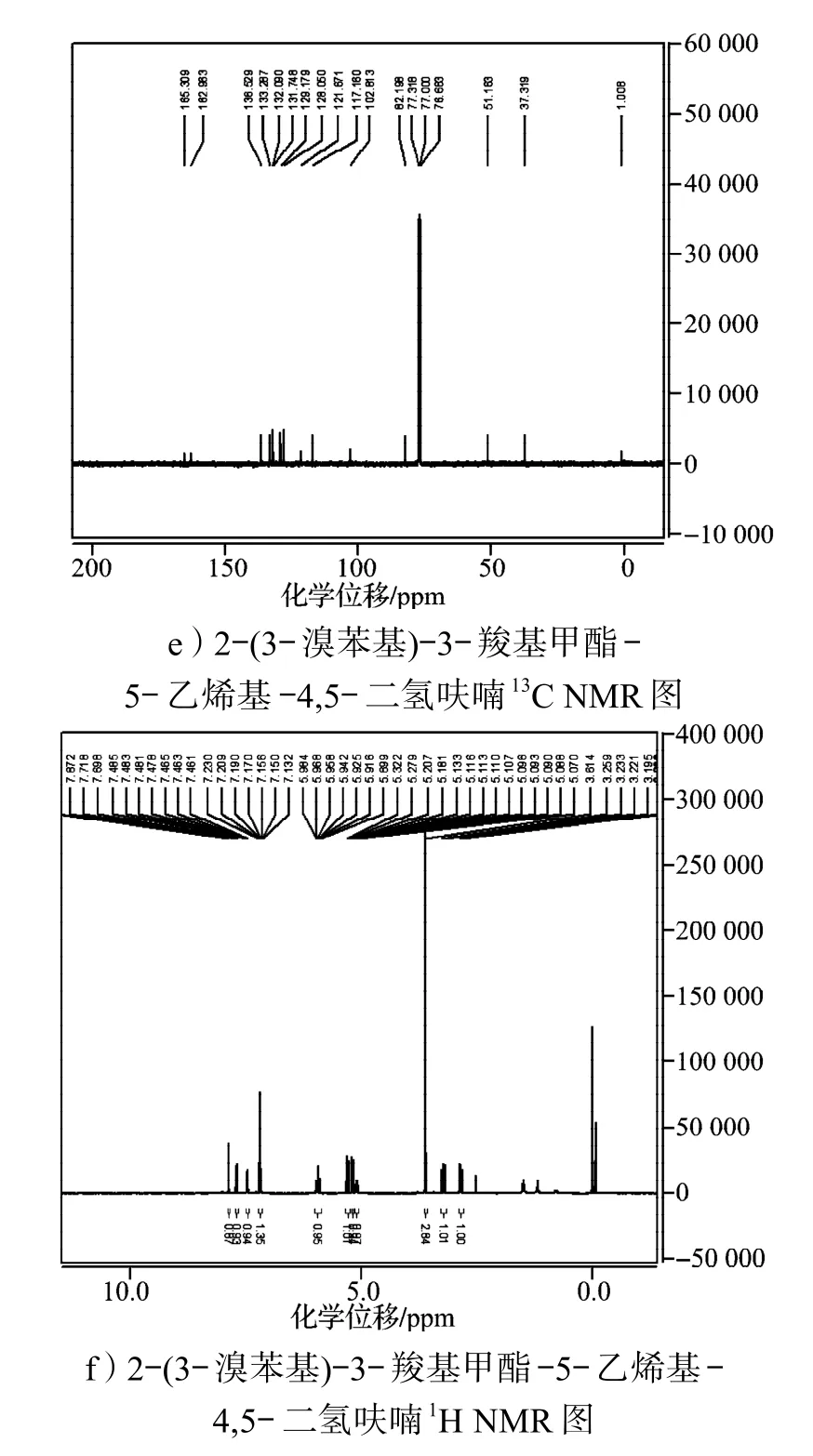
圖2 部分呋喃環衍生物的核磁共振譜圖Fig.2 The NMR spectra of part of furan derivatives
2 結果與討論
2.1 試驗條件的優化
以0.75 mmol 苯甲酰乙酸甲酯1a與0.5 mmol 烯丙基二碳酸酯2為反應底物來考察它們在四(三苯基膦)鈀(Pd(PPh3)4)催化條件下得到的呋喃環衍生物。以圖3所示的反應對試驗條件進行優化。
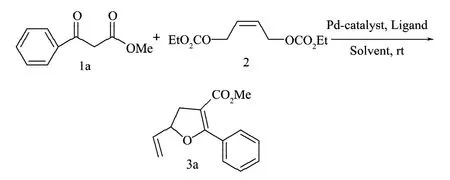
圖3 2-苯基-3-羧基甲酯-5-乙烯基-4,5-二氫呋喃的合成Fig.3 The synthesis of 2-phenyl-3-carboxylate-methyl-5-vinyl-4,5-dihydrofuran
為確證其它因素(如配體、溶劑等)對試驗結果的影響,現對其進行篩選(如表1)。首先是配體種類的篩選,如1,2-雙(二苯基膦)乙烷(DPPE)、1,1’-雙(二苯基膦)二茂鐵(DPPF)、1,4-雙(二苯基膦)丁烷(DPPB)、1,3-雙(二苯基膦)丙烷(DPPP)和三苯基膦(PPh3);其次是溶劑種類的篩選,如THF,MeOH,EDC,Toluene,1,4-Dioxane和DCM;最后是金屬催化劑和配體的用量確定等。
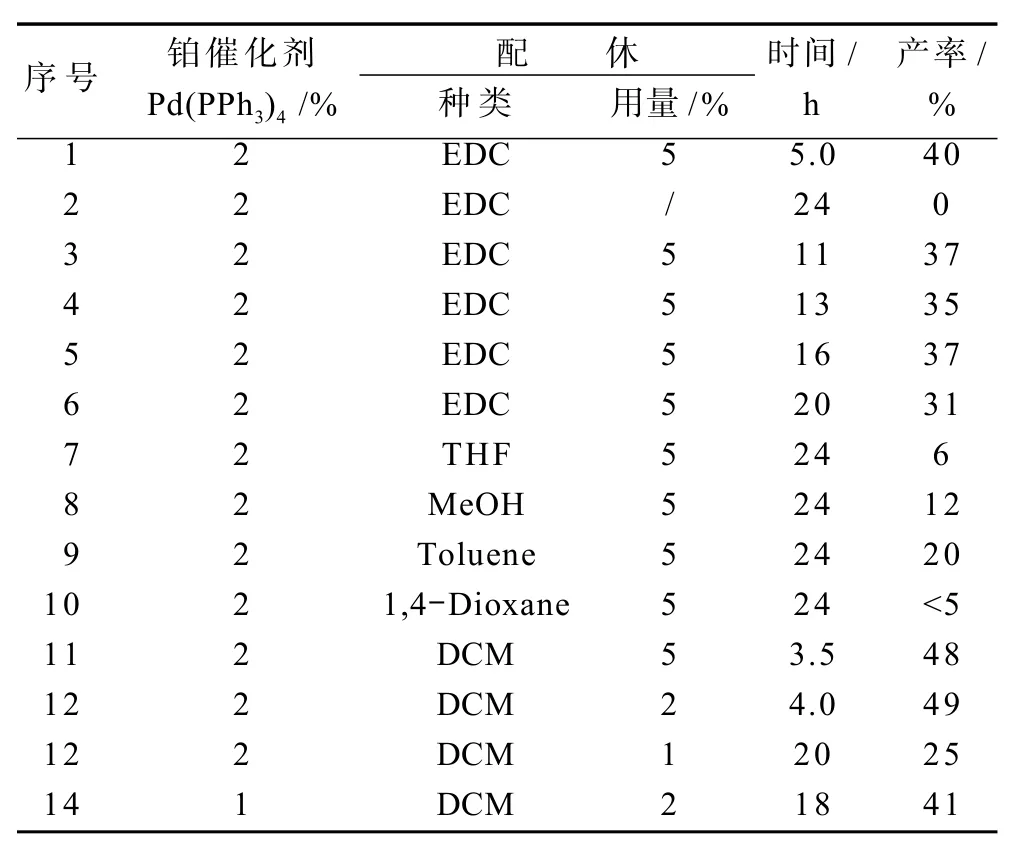
表1 合成條件的優化Table 1Optimization of synthetic conditions
通過觀察金屬催化劑與配體作用時發現(表1,序號1~2),Pd(PPh3)4單獨作用下不能催化合成呋喃環,只有在與配體共同作用時才能使此反應進行,生成呋喃衍生物。配體篩選時發現(表1,序號1,3~6),DPPE較其他配體作用時有較好的反應活性和產率,其他配體如DPPF,DPPB,DPPP和PPh3與金屬催化劑共同作用下雖都能促進反應進行,生成呋喃環,但其反應速度都較DPPE作用時慢,生成物的產率也較低。在其它條件不變的條件下,以DPPE為配體進行溶劑篩選時發現(表1,序號1,7~11),分別以DCM和EDC作為反應溶劑時,反應情況較好,反應體系較純凈,以THF,MeOH,Toluene、1,4-Dioxane作為反應溶劑時,雖有產物生成,但是產率卻不是很高,尤以1,4-Dioxane作為溶劑時,反應體系較復雜且產率極低(小于5%)。但是以DCM與EDC作為反應溶劑時,DCM較EDC有較好的反應活性與反應速率,且DCM作為溶劑時只需用4A分子篩簡單除水即可,不需經過嚴格的去水處理。總的來說,不含氧溶劑(如DCM,EDC,Toluene)的反應速率較含氧溶劑(如MeOH,1,4-Dioxane)快。確定反應的加入量進行時發現(表1,序號11~14):以烯丙基二碳酸酯的物質的量為用量標準,當金屬催化劑Pd(PPh3)4用量為標準用量的2%、配體DPPE用量為標準用量的2%時,達到最優用量,其它比例的用量造成原料的浪費或使反應不徹底。對各物質種類及其用量進行確定后,再對條件溫度進行優化。將溫度條件分別設為:較低溫度-15℃與0℃;較溫和溫度,即10℃與25℃;二氯甲烷沸點溫度,40℃。試驗結果發現,在-15 ℃及0℃條件下,反應速率慢于其他3個溫度條件下的反應速率,且生成物產率較低,而在10℃,25℃與40 ℃溫度條件下的反應速率相差不大,且各生成物產率也相差不大,故將反應溫度條件設為室溫。
通過上述對各條件的篩選,確定此反應的最優化條件如下:底物-酮酯1a為0.75 mmol、底物烯丙基二碳酸酯2為0.5 mmol、催化劑四(三苯基膦)鈀為0.01 mmol、配體1,2-雙(二苯基膦)乙烷為0.01 mmol、溶劑二氯甲烷為1.5 mL、室溫。
2.2 系列呋喃衍生物的合成

圖4 系列呋喃衍生物的合成Fig.4 The synthesis of a series of furan derivatives
通過對表2分析可知:苯環上取代基的種類對反應產率有較大的影響,當苯環上增加取代基后,其反應產率有所增加。為了觀察電子效應對反應結果的影響,本課題組研究了苯環上含有給電子基團的甲氧基取代基及吸電子基團的氟與氯取代基,觀察的結果表明,電子效應對反應的影響較大,吸電子基團較給電子基團有較高的產率,尤其是含有間溴取代基的,其產率達到83%。分析苯環上取代基位置對反應的影響時發現:不論是吸電子基團取代基(序號5~6與序號7~9)還是給電子基團取代基(序號2~3),苯環上同一位點不同的取代基(序號2,5,9;序號3,8與序號6~7)對試驗結果有較大影響,同一取代基(序號2~3;序號5~6與序號7~9)不同的位點對試驗結果亦有較大的影響。從表2可看出,不論是給電子基團還是吸電子基團,反應速率都較快,能在2.5~6.0 h內反應完全,并達到較高的產率。
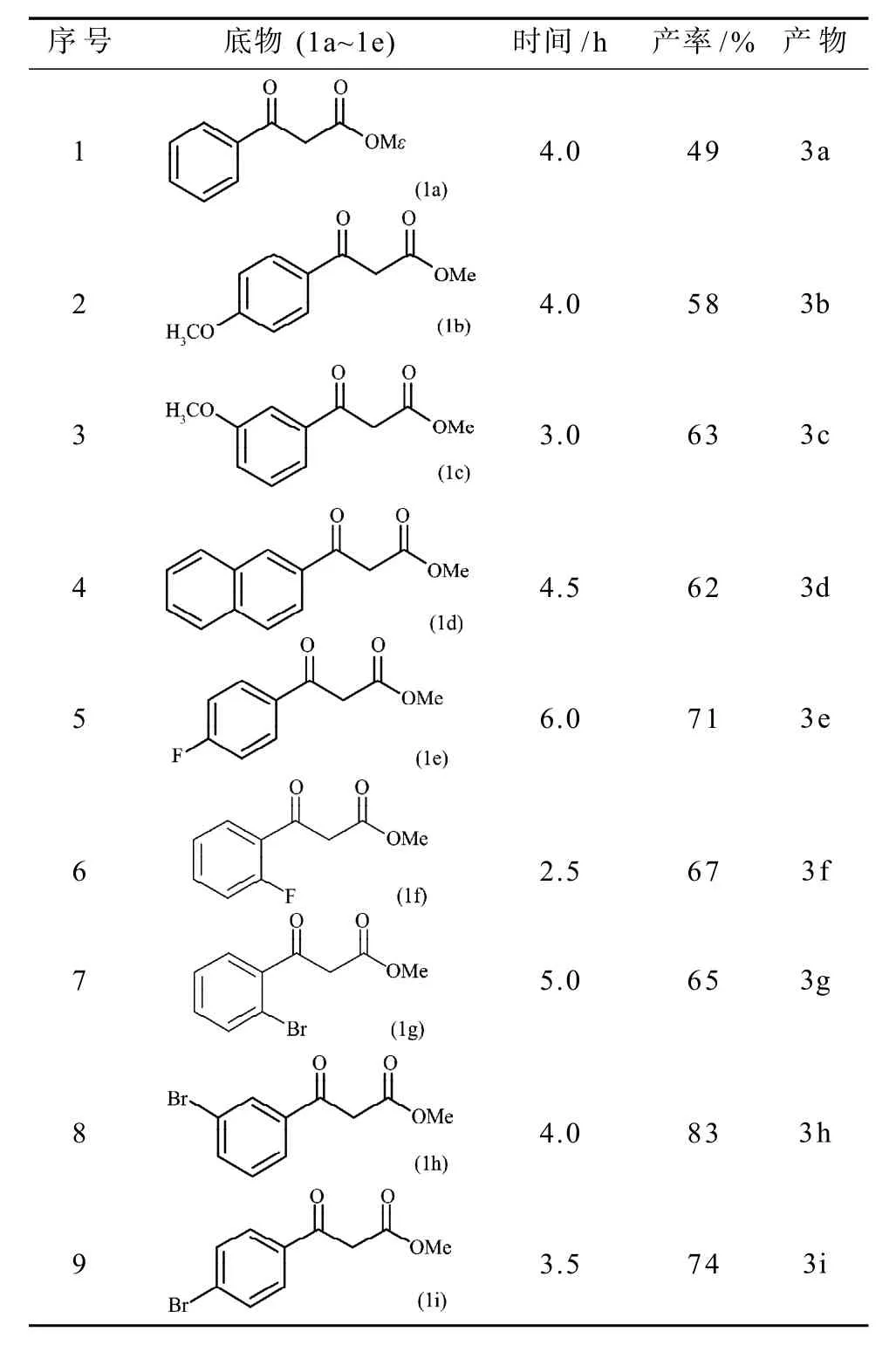
表2 系列呋喃衍生物的合成Table 2The synthesis of a series of furan derivatives
2.3 反應機理的探討
依據文獻[19]類似機理的報導與對表1和2的試驗結果分析,提出了下面的可能機理:首先烯丙基二碳酸酯在零價鈀作用下脫除單碳酸酯負離子生成配合物I,緊接著單碳酸酯負離子進攻-酮酯生成互變異構負離子IIA和IIB,然后碳負離子IIA通過分子間的C-烯丙基化反應進攻配合物Ⅰ生成中間體III,中間體III繼續通過和零價鈀配位、脫碳酸酯,并通過分子內的O-烯丙基化反應選擇性地得到呋喃環衍生物3a。其可能的反應機理如圖5所示。
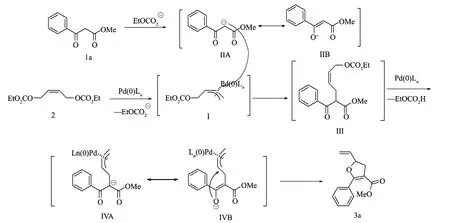
圖5 反應機理的探討流程圖Fig.5 The flowchart for the reaction mechanism investigation
3 結語
由于含呋喃骨架的化合物廣泛存在于天然產物中,并具有多種生物活性,如:抗癌、抗菌等,此類衍生物的合成引起眾多從事有機合成者的關注。利用-酮酯與對稱的烯丙基二碳酸酯為原料,以烯丙基二碳酸酯的物質的量為用量標準,在以占標準用量2%的四(三苯基膦)鈀與占標準用量2%的1,2-雙-(二苯基膦)乙烷的作用下,通過分子間的C-烯丙基化反應與分子內的O-烯丙基化反應在一定條件下以中等的產率得到一系列的含有呋喃骨架的2,3-二取代-5-乙烯基-4,5-二氫呋喃衍生物。試驗結果表明:電子效應對實驗結果有較大的影響,吸電子基團較給電子基團有較高的活性及產率;不同的取代基對試驗結果有影響,取代基的位置對反應結果的影響也較大。此方法具有催化用量少、反應條件溫和及操作簡便等優點,該研究為后續正在進行的的手性呋喃環衍生物的合成提供了依據,也為呋喃類藥物的合成與研究提供了新的思路與方法。
[1]Rahmathullah S M,Hall J E,Bender B C,et al. Prodrugs for Amidines:Synthesis and Anti-Pneumocystis Carinii Activity of Carbamates of 2,5-Bis(4-Amidinophenyl)Furan [J]. Journal of Medicinal Chemistry,1999,42(19):3994-4000.
[2]Florio R D,Rizzacasa M A. Synthesis of 2,2-Disubstituted Furanoid Natural Products:Total Synthesis of Sphydrofuran[J]. Journal of Organic Chemistry,1998,63 (23):8595-8598.
[3]Anbazanagn M,Boykin D W. A Facile Synthesis of the Prodrug 2,5-Bis(4-0-Methoxyamidinophenyl)Furan and Analogs[J]. Heterocyclic Communications,2003,9(2):117-118.
[4]Kraft P,Baigrowicz J A,Denis C,et al. Odds and Trends:Recent Developments in the Chemistry of Odorants[J]. Angewandte Chemie International Edition,2000,39(17):2980-3010.
[5]Nizamuddin,Madhuri G,Kumar S M. Synthesis and Fungicidal Activity of Some Furan Containing Azoles[J]. Journal of Scientific & Industrial Research,1999,58(12):1017-1020.
[6]Sugimura H,Osumi K,Kodaka Y,et al. Stereoselective Synthesis of 2’-Deoxy--D-Threopentofurano Nucleosides by the NBS-Promoted Coupling Reaction of Thioglycosides with Silylated Heterocyclic Bases[J]. Journal of Organic Chemistry,1994,59(25):7653-7660.
[7]Tanimori S,Kirihata M. Palladium-Catalyzed Approach to Substituted Dihydrofurans,Cyclopenta[b]Furans and Tetrahydrobenzofurans[J]. Synthesis,2007(1):39-44.
[8]Bowman R K,Johnson J S. Nickel-Catalyzed Rearrangement of 1-Acyl-2-Vinylcyclopropanes:A Mild Synthesis of Substituted Dihydrofurans[J]. Organic Letters,2006,8(4):573-576.
[9]Tanimori S,Kato Y,Kirihata M. Simple Preparation of New Functionalized Furan Derivatives Via Sequential C-C and C-O Bond Formation Mediated by Palladium-Phosphine Catalyst[J]. Synthesis,2006(5):865-869.
[10]Ohno A,Yamane M,Hayashi T,et al. Preparation and Use of Chiral Ferrocenylphosphines Containing New Alkyl Substituents on the Ferrocenylmethyl Position[J]. Tetrahedron,1995,6(10):2495-2502.
[11]Murakami H,Matsui Y,Ozawa F,et al. Cyclodehydration of Cis 2-Butene-1,4-Diol with ActiveMethylene Compounds Catalyzed by a Diphosphinidenecyclobutene-Coordinated Palladium Complex[J]. Journal of Organometallic Chemistry,2006,691(4):3151-3156.
[12]Sieller B,Bruneau C,Dixneuf P H. Novel Ruthenium-Catalysed Synthesis of Furan Derivatives Via Intramolecular Cyclization of Hydroxy Enynes[J]. Chemical Communications,1994(4):493-494.
[13]Hashmi A S K,Sinha P. Gold Catalysis:Mild Condition for the Transformaion of Alkynyl Epoxides to Furans[J]. Adv. Synth. Catal.,2004,346(4),432-438.
[14]Stergiou A,Bariotaki A,Kalaitzakis D,et al. Oxone-Mediated Oxidative Cleavage of-Keto Esters and 1,3-Diketones to -Keto Esters and 1,2-Diketones in Aqueous Medium[J]. The Journal of Organic Chemistry,2013,78 (14):7268-7273.
[15]Liu X,Cheng R,Zhao F F,et al. Direct-Acyloxylation of Enamines Via PhIO-Mediated Intermolecular Oxidative C-O Bond Formation and Its Application to the Synthesis of Oxazoles[J]. Organic Letters,2012,14(21):5480-5483.
[16]Russo A,Galdi G,Croce G,et al. Highly Enantioselective Epoxidation Catalyzed by Cinchona Thioureas:Synthesis of Functionalized Terminal Epoxides Bearing a Quaternary Stereogenic Center[J]. Chemistry a European Journal,2012,18(20):6152-6157.
[17]Morgen M,Bretzke S,Li P F,et al. Stereodivergent Synthesis of 1,3-Syn-and-Anti-Tetra-Hydropyrimidinone-S[J]. Organic Letters,2010,12(20):4494-4497.
[18]Hodgson D M,Stefane B,Miles T J,et al. Organolithium-Induced Alkylative Ring Opening of Aziridines:Synthesis of Unsaturated Amino Alcohols and Ethers[J]. The Journal of Organic Chemistry,2006,71(22):8510-8515.
[19]Hu Z J,Li Y G,Liu K,et al. Bis(Perfluoroalkyl) Phosphino-Oxazoline:A Modular,Stable,Strongly -Accepting Ligand for Asymmetric Catalysis[J]. The Journal of Organic Chemistry,2012,77(18):7957-7967.
(責任編輯:申劍)
The Synthesis of 2,3-Bisubsituted-5-Vinyl-4,5-Dihydrofuran
Xiao Li,Zhou Zhipeng,Wang Haifei,Hu Shunqin
(School of Packaging and Materials Engineering,Hunan University of Technology,Zhuzhou Hunan 412007,China)
In order to solve the problems of adding expensive metal catalyst or alkali to synthesize furan ring,taking symmetrical allyl carbonate as the dosage standard,applies-ketoester and symmetrical allyl carbonate as raw materials to synthesize 2,3-bisubsituted-5-vinyl-4,5-dihydrofuran derivatives under the combined catalysis of the standard 2% tetrakis (triphenylphosphine)palladium catalyst and of the standard 2% 1,2-bis(diphenylphosphino)ethane. The experimental results show that the reaction has high yields with the electron withdrawing groups. By changing the substituents,obtains 9 different substituted 2,3-bisubsituted-5-vinyl-4,5-dihydrofuran derivatives. All products are confirmed by1H and13C NMR.
-ketoester;allyl carbonate;dihydrofuran
O626.11
A
1673-9833(2015)01-0102-07
2014-09-03
國家自然科學青年基金資助項目(21202042),湖南省自然科學青年基金資助項目(13JJ4090)
肖利(1989-),男,湖南洞口人,湖南工業大學碩士生,主要研究方向為有機催化合成,E-mail:5939702712@qq.com
胡舜欽(1967-),男,湖南桃江人,湖南工業大學教授,博士,主要從事分析化學方面的研究,E-mail:1328235513@qq.com
10.3969/j.issn.1673-9833.2015.01.019
