血塞通分散片指紋圖譜研究
2015-08-10 09:31:38段紅吉來國防楊光梅
云南中醫學院學報 2015年6期
段紅吉,張 洪,來國防,楊光梅
(1.云南植物藥業有限公司,云南 昆明650109;2.云南省食品藥品檢驗所,云南 昆明650011)
血塞通分散片主要成分是從三七中提取的三七總皂苷和相應量輔料,精制而成(規格:50mg/片)。血塞通分散片主要成分為三七總皂苷(三七皂苷R1、人參皂苷Rg1、人參皂苷Re、人參皂苷Rb1、人參皂苷Rd)[1-2]。
血塞通分散片主要用于散瘀止血,消腫定痛;用于咯血,吐血,便血,崩漏,外傷出血,胸腹刺痛,跌撲腫痛,能活血化瘀、止痛。血塞通分散片能抑制由ADP 引起的家兔血小板聚集,擴張腦血管,使腦血流量增加,也具有抗血栓和抗凝血作用[3]。
指紋圖譜是評價中藥及其制劑質量穩定的有效方法,通過多成分的特征峰及多維的數據角度進行綜合分析揭示血塞通分散片的指紋圖譜特征[4-6],為血塞通分散片的質量控制提供新的參考方法。
1 材料與方法
1.1 材料與試劑
血塞通分散片(10 批批號分別為:20150501、20150502、20150503、20150601、20150602、20150603、20150605、20150606、20150607、20150608),樣品由云南植物藥業有限公司提供。
三七總皂苷對照品(含三七皂苷R16.9%、人參皂苷Rg128%、人參皂苷Re 3.8%、人參皂苷Rb129.7%、人參皂苷Rd 7.3%),購自中國食品藥品檢定研究院,批號:110870-201002。純化水為屈臣氏純化水;乙腈(色譜純),甲醇(色譜純),其余試劑為分析純。
1.2 儀器與設備
Agilent1260 液相色譜儀(配有二極管陣列檢測器、四元梯度泵、在線脫氣裝置、自動進樣器),Chem-Station 工作站(Agilent 科技有限公司),用十八烷基硅烷鍵合硅膠為填充劑的色譜柱作為分析柱[7-8]。
1.3 對照品與供試品制備
1.3.1 對照品溶液的制備
取三七總皂苷對照提取物適量,精密稱定,加70%甲醇使溶解,并稀釋制成每1mL 含2.5mg 的溶液,即得。
1.3.2 供試品溶液的制備
本制劑為三七總皂苷加適量賦形劑制成的片劑。因《中華人民共和國藥典》2010 版一部“三七”項藥材中皂苷類成分含量測定時選用甲醇為提取溶劑,“三七總皂苷”項提取物指紋圖譜時選用70%甲醇為提取溶劑,且本制劑中多數輔料在乙醇中不易溶解,故選擇此3 種溶劑進行提取考察。即:取重量差異項下的同一批次樣品,研細,稱取適量(約相當于三七總皂苷25mg)3 份,各置10mL 量瓶中,分別精密加入甲醇、乙醇、70%甲醇至刻度,稱定重量,超聲處理(功率250W,頻率33kHz)20min,放冷,再稱定重量,并用相應溶劑補足減失的重量,作為供試品溶液。同時,考察了3 個不同提取時間對本品指紋圖譜的影響。即:取重量差異項下的同一批次樣品,研細,稱取適量(約相當于三七總皂苷25mg)3 份,各置10mL 量瓶中,加入甲醇至刻度,稱定重量,分別超聲提取(功率250W,頻率33kHz)10,20,30min 后,放冷,再稱定重量,并用甲醇補足減失的重量,作為供試品溶液。按1.4 項的色譜條件,分別吸取對照品溶液和供試品溶液各20μL 進樣,記錄色譜圖,測定其相似度,結果見表1。
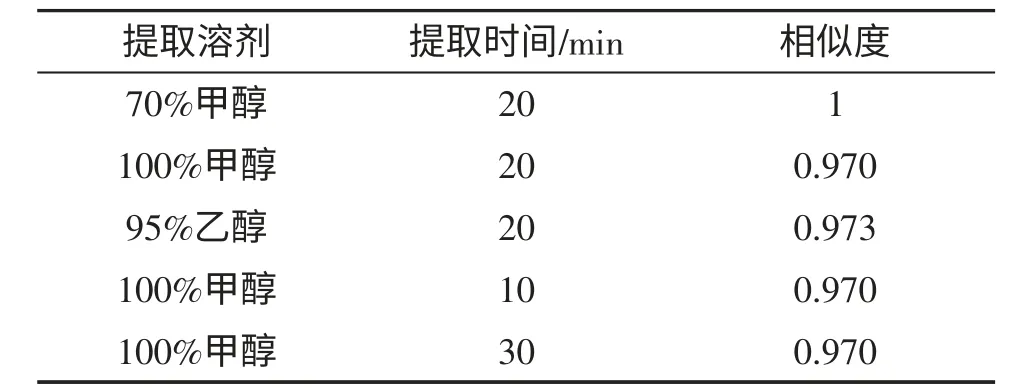
表1 提取因素考察
結果表明3 種提取溶劑及提取時間,對圖譜無明顯影響。故選擇70%甲醇為提取溶劑,超聲時間為10min,并將此條件列入正文。
1.3.3 陰性對照溶液的制備
按處方比例及制法,制備缺三七總皂苷對照藥品。再按上述所得供試品溶液制備方法制成缺三七總皂苷的陰性對照溶液。
1.4 色譜條件與系統適用性試驗
1.4.1 色譜柱、檢測波長的選擇
參照《中華人民共和國藥典》2010 版一部“三七總皂苷”指紋圖譜項,選用十八烷基硅烷鍵合硅膠為填充劑的色譜柱作為分析柱,檢測波長為203nm。
1.4.2 流動相比例的選擇
照《中華人民共和國藥典》2010 版一部“三七總皂苷”指紋圖譜項,選擇以乙腈為流動相A,以水為流動相B,進行梯度分析。以分離度、峰的對稱性、保留時間等為評價指標。結果表明以下表流動相比例較好,見表2。

表2 流動相比例的選擇
1.4.3 系統適用性試驗
見圖1-3,根據供試品的HPLC 圖譜,色譜柱的理論塔板數以人參皂苷Rg1峰計均不低于6 000。人參皂苷Rg1、人參皂苷Re 的分離度均大于1.5。
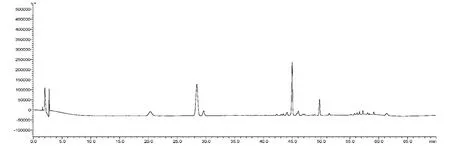
圖1 對照品HPLC 圖譜
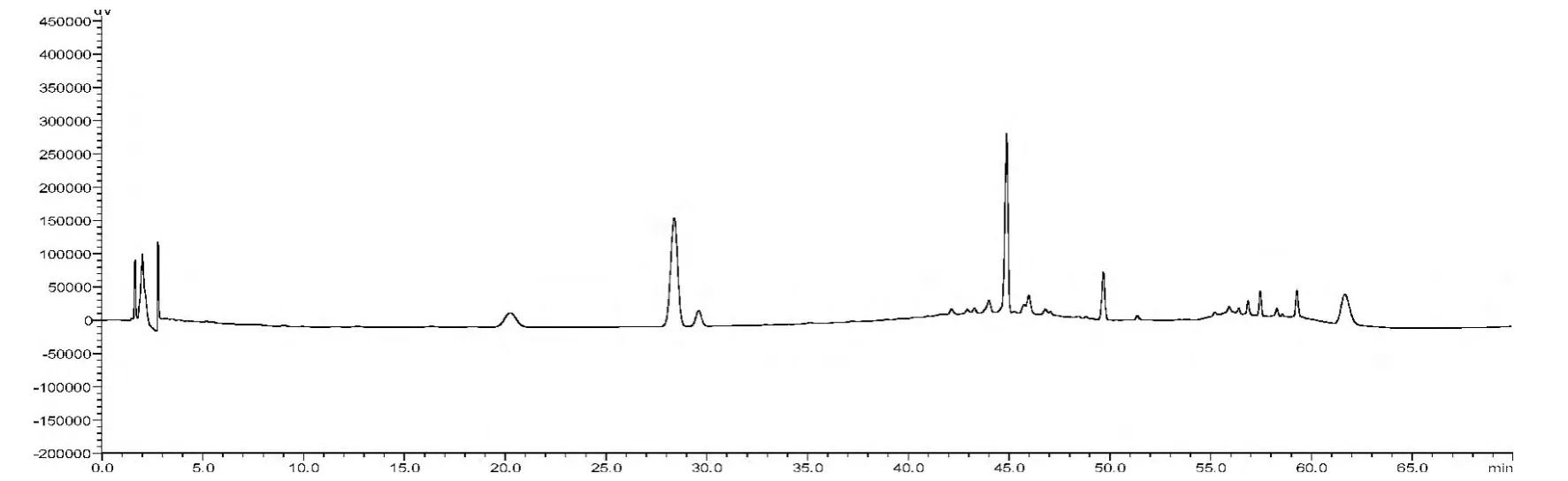
圖2 供試品HPLC 圖譜
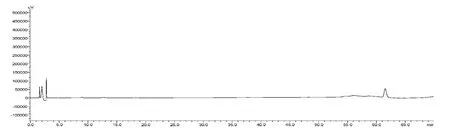
圖3 陰性對照藥品HPLC 圖譜
2 指紋圖譜及技術參數
取10 批不同生產批次的血塞通分散片,照2.2所得方法進行處理,并按1.4 項的色譜條件,分別吸取對照品溶液和供試品溶液各20μL 進樣,記錄色譜圖,記為S1-S11,其中S1對照品色譜,S2-S11分別為批號20150501、20150502、20150503、20150601、20150602、20150603、20150605、20150606、20150607、20150608 的血塞通分散片圖譜,見圖4。利用《中藥色譜指紋圖譜相似度評價系統2004A 版》軟件,采用中位數法,時間窗為0.1,對圖譜信息進行相似度評價及共有峰的共有圖譜信息擬合,得到血塞通分散片的HPLC 指紋圖譜,然后確定共有峰[9]。計算單峰面積占總峰面積的百分比,并以保留時間和峰面積較為平穩的人參皂苷Rg1色譜峰作為參比峰,計算各共有指紋峰的相對保留時間和相對峰面積的值。
2.1 指紋圖譜
10 批血塞通分散片供試品生成共有峰32 個。其中,對照品與供試品生成共有峰29 個。選定其中5 個共有峰作為共有指紋峰,指紋峰的選擇原則是匹配度為100%,相對峰面積比值RSD≤10%,相對峰面積比值≥0.1[10]。經單峰定位后,確定此5 個峰,按保留時間由小到大,依次分別為三七皂苷R1、人參皂苷Rg1、人參皂苷Re、人參皂苷Rb1和人參皂苷Rd,為三七總皂苷中的主要有效成分,見圖5-6。
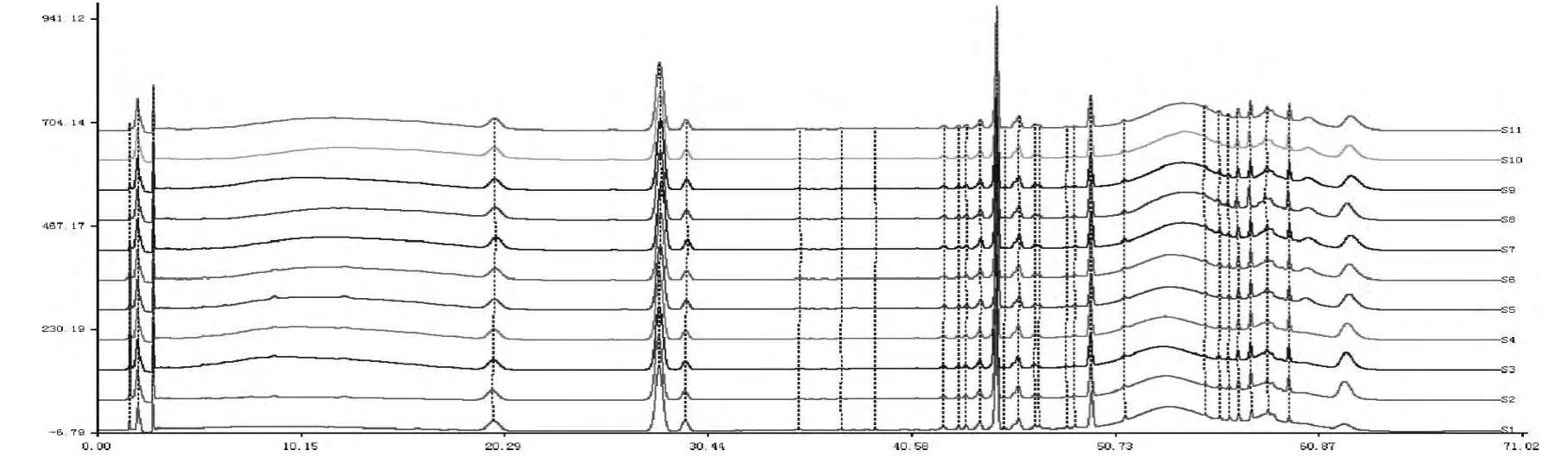
圖4 對照品及10 批供試品色譜圖
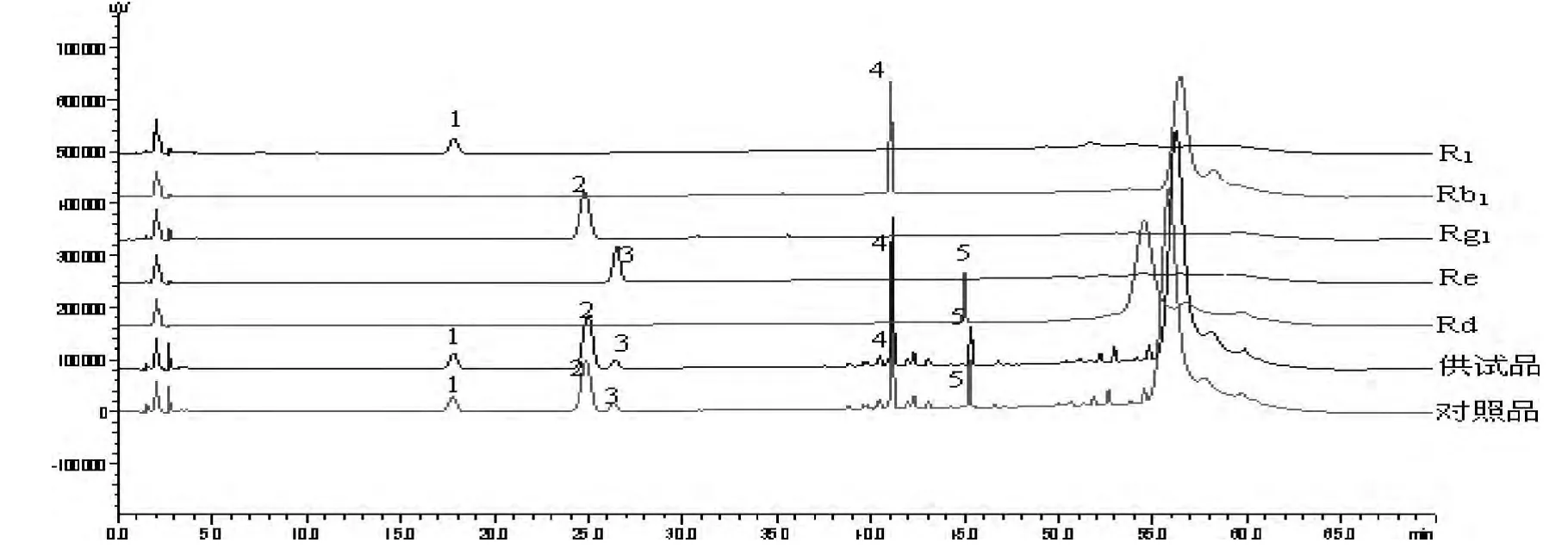
圖5 單峰定位結果
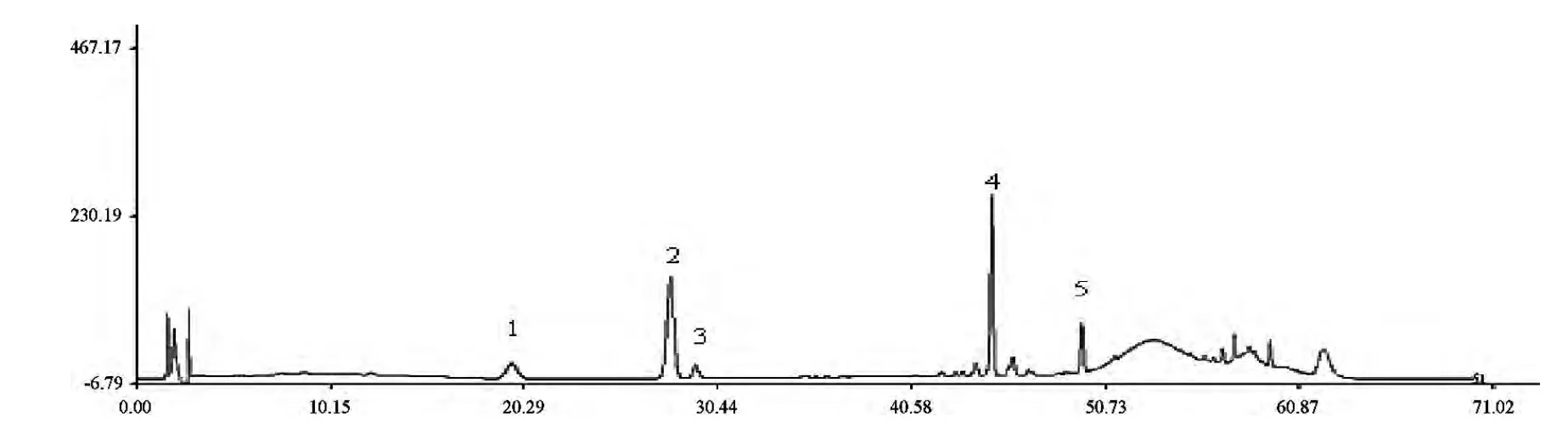
圖6 共有指紋峰圖譜
2.2 共有峰的峰號及保留時間
1 號峰,保留時間:19.774,為三七皂苷R1
2 號峰,保留時間:28.029,為人參皂苷Rg1
3 號峰,保留時間:29.335,為人參皂苷Re
4 號峰,保留時間:44.810,為人參皂苷Rb1
5 號峰,保留時間:49.503,為人參皂苷Rd以保留時間和峰面積較為平穩的人參皂苷Rg1色譜峰作為參比峰,計算各共有指紋峰峰面積的比值,見表3。
結果表明,5 個峰的相對峰面積比值RSD 小于5%,峰面積比值均在允許范圍(準許誤差范圍是利用《中藥色譜指紋圖譜相似度評價系統2004A 版》軟件所得)內,5 個指紋峰的相對保留時間一致性較好,指紋圖譜具有相似度較高的共有峰。
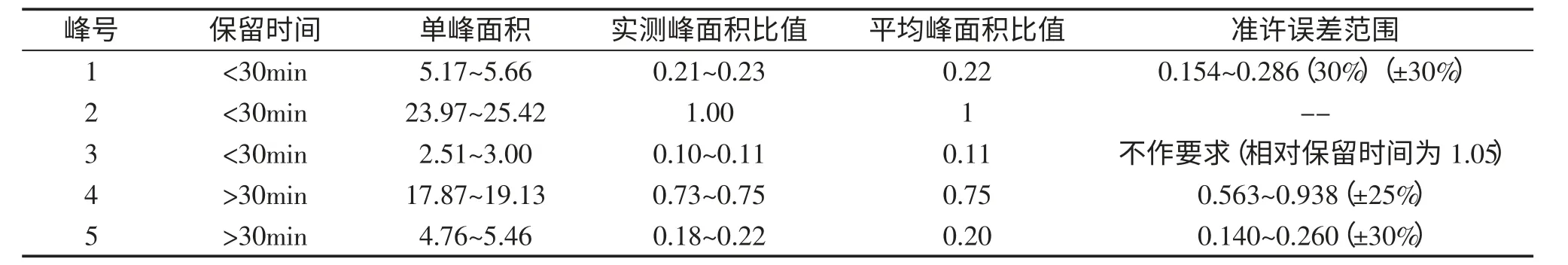
表3 共有指紋峰峰面積的比值
2.3 非共有峰
非共有峰面積占總峰面積10%~15%.
3 方法學考察
3.1 專屬性試驗
分別精密吸取混合對照品溶液、供試品溶液、陰性對照溶液各20μL,進樣,記錄色譜圖,見圖1-3。在該色譜條件下的分離效果較好,對照品圖譜與供試品圖譜,相似度為0.98。陰性樣品在于對照品及供試品出峰相應的保留時間處未見明顯色譜峰,表明本品中輔料成分對三七皂苷R1、人參皂苷Rg1、人參皂苷Re、人參皂苷Rb1和人參皂苷Rd 的測定無干擾。
3.2 精密度
取同一批次血塞通分散片,研細,稱取適量(約相當于三七總皂苷25mg),精密稱定,照2.2 所得方法進行處理,并按1.4 項的色譜條件,連續進樣5次,每次20μL。記錄色譜圖,記為S1-S5。利用《中藥色譜指紋圖譜相似度評價系統2004A 版》軟件,對圖譜信息進行相似度評價,計算相似度[11]。以人參皂苷Rg1色譜峰作為參比峰,計算各共有指紋峰的相對保留時間和相對峰面積的RSD 值,結果見表4。
3.3 重復性
取重量差異項下本品,研細,稱取適量(約相當于三七總皂苷25mg)6 份,照2.2 所得方法進行處理,并按1.4 項的色譜條件,吸取供試品溶液各20μL進樣,記錄色譜圖,記為S1-S6。利用《中藥色譜指紋圖譜相似度評價系統2004A 版》軟件,采用中位數法,時間窗為0.1,對圖譜信息進行相似度評價,計算相似度。以保留時間和峰面積較為平穩的人參皂苷Rg1色譜峰作為參比峰,計算各共有指紋峰的相對保留時間和相對峰面積的RSD 值,結果見表5。
3.4 穩定性
取同一批次血塞通分散片,研細,稱取適量(約相當于三七總皂苷25mg),精密稱定,照2.2 所得方法進行處理,并按1.4 項的色譜條件,分別于0,2,4,8,12,16 h 精密吸取同濃度供試品溶液20μL 進樣,記錄色譜圖,記為S1-S6。利用《中藥色譜指紋圖譜相似度評價系統2004A 版》軟件,采用中位數法,時間窗為0.1,對圖譜信息進行相似度評價,計算相似度。以保留時間和峰面積較為平穩的人參皂苷Rg1色譜峰作為參比峰,計算各共有指紋峰的相對保留時間和相對峰面積的RSD 值,結果見表6。

表4 精密度考察
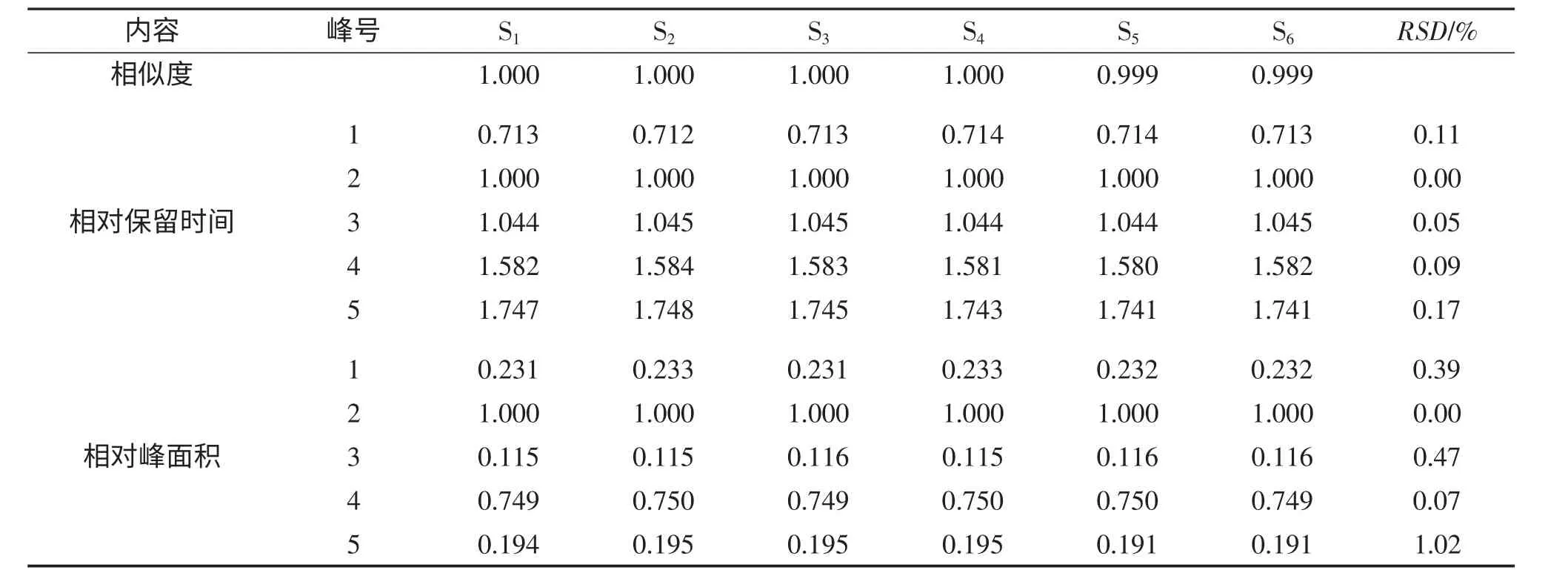
表5 重復性測定結果
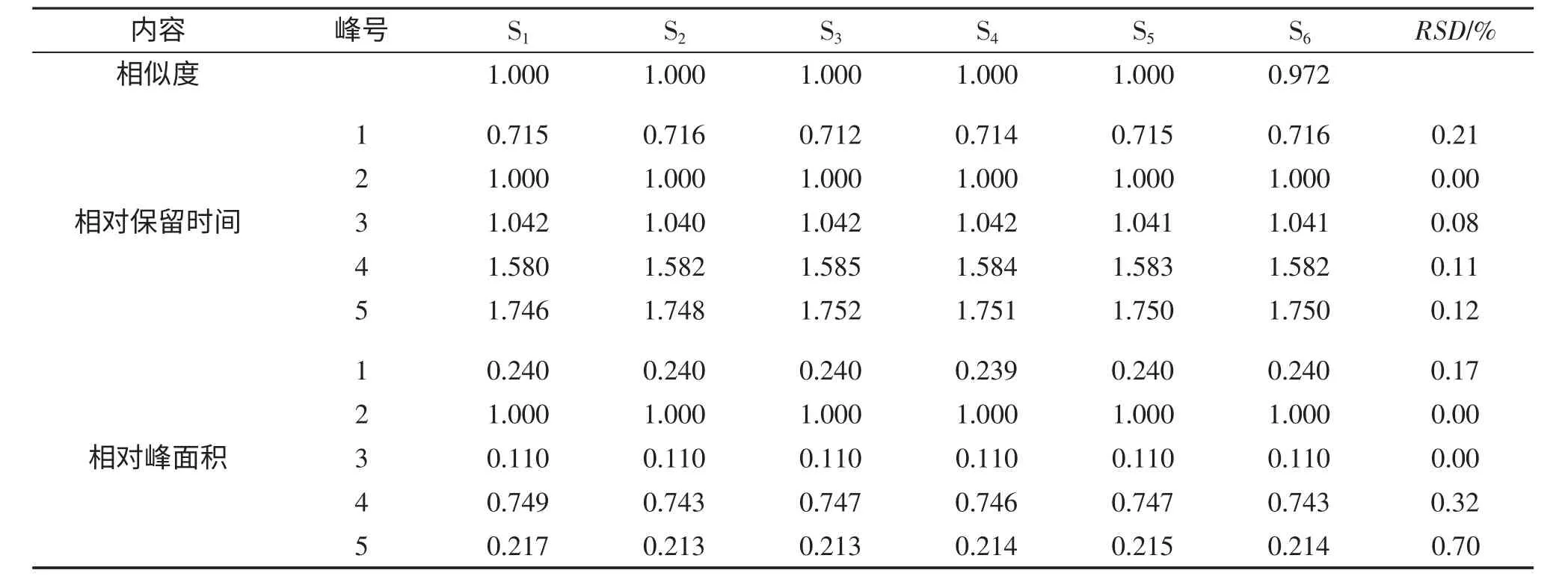
表6 穩定性考察
4 總結
本文以三七皂苷R1、人參皂苷Rg1、Re、Rb1、Rd峰為參照峰,建立了血塞通分散片HPLC 指紋圖譜。應用“中藥色譜指紋圖譜超信息特征評價系統”軟件對血塞通分散片HPLC 指紋圖譜定性、定量信息進行多維的數據分析。采用雙定性相似度(SF 和SF’)和雙定量相似度(C 和P)來評價血塞通注射液指紋圖譜的化學成分分布比例特征和含量分布特征。評價出10 批血塞通注射液質量完全合格。本實驗所建立的血塞通注射液指紋圖譜具有良好的精密度和重現性,結合雙定性雙定量相似度評價法能較好地控制其質量[12-15]。
[1] 國家藥典委員會. 中華人民共和國藥典(一部)[M]. 北京:化學工業出版社,2005:10.
[2] 丁婷. 血塞通分散片制劑工藝的研究[J]. 長沙醫學院學報,2014,12(1):29-36.
[3] 張曉青,張陽根. 血塞通分散片對心腦保護作用的實驗研究[J]. 中國現代醫學雜志,2005,15(10):1503-1505.
[4] 楊雪梅,劉旭,嚴軼琛,等. 三七藥材指紋圖譜的研究[J].第一軍醫大學學報,2004,24(12):1410 -1411.
[5] 黎瓊紅,張國剛. 三七藥材的指紋圖譜[J]. 沈陽藥科大學學報,2006,23(4):229 -232.
[6] 劉曉麗,周清,孫國祥. 血塞通注射液HPLC 數字化指紋圖譜研究[J]. 遼寧中醫雜志,2012,39(11):2238-2241.
[7] 代龍. HPLC 法測定血塞通分散片中三七皂苷R1及人參皂苷Rg1、Rb1的含量[J]. 奇魯藥事,2009,28(6):333-335.
[8] 汪存存. 血塞通分散片的含量測定方法分析[J]. 國際醫藥衛生導報,2008,14(6):58-63.
[9] 王芳,張賀,葛平,等. 基于高效液相色譜技術的中藥制劑指紋圖譜構建及應用現狀[J]. 中國基層醫藥,2014,21(16):2548-2550.
[10] 丁紅霞,楊靜,吳宗耀,等. 中藥質量控制中色譜指紋圖譜技術探究[J]. 河南中醫,2013,33(7):1155-1156.
[11] 許丹萍. 中藥色譜指紋圖譜相似度的計算[J]. 數學學習與研究,2012(13):119.
[12] 李昕,王月茹,馬利,等. 中藥色譜指紋圖譜在中藥質量評價中的應用[J]. 生物醫學工程學雜志,2012,29(1):192-196.
[13] 田立權. 中藥指紋圖譜的主要技術分析手段概論[J]. 民營科技,2011(2):98.
[14] 任非,智麗敏,付穎,等. 中藥指紋圖譜色譜技術及其在質量控制中的應用與研究[J]. 河北醫藥,2010,32(21):3075-3078.
[15] 梁逸曾,王兵,曾茂茂,等.色譜指紋圖譜與中藥質量控制[J].世界科學技術—中醫藥現代化,2010,12(1):94-98.
