固載型雜化微球模擬催化氧化脫硫性能
2015-08-19 06:46:46宋少飛周福林弓巧娟沈淑坤
化工進(jìn)展 2015年4期
關(guān)鍵詞:催化劑實(shí)驗(yàn)
宋少飛,周福林,弓巧娟,沈淑坤
(1運(yùn)城學(xué)院應(yīng)用化學(xué)系,山西 運(yùn)城 044000;2陜西師范大學(xué)材料科學(xué)與工程學(xué)院,陜西 西安 710062)
近年來(lái),環(huán)境污染的問(wèn)題日趨嚴(yán)重,已經(jīng)引起了世界各國(guó)的普遍關(guān)注。相關(guān)研究表明,機(jī)動(dòng)車(chē)尾氣排放是造成空氣質(zhì)量下降的主要因素。為了提高油品的質(zhì)量,從根本上降低尾氣排放帶來(lái)的危害,國(guó)際上陸續(xù)制定了嚴(yán)格的含硫尾氣排放標(biāo)準(zhǔn)[1-2]。與此相應(yīng),燃料油脫硫技術(shù)也應(yīng)運(yùn)而生,如加氫脫硫已實(shí)現(xiàn)工業(yè)化應(yīng)用。但加氫脫硫技術(shù)對(duì)大分子的雜環(huán)含硫化合物(如二苯并噻吩及其衍生物)無(wú)法實(shí)現(xiàn)有效脫除,不能達(dá)到深度脫硫的目的。因此,燃油深度脫硫技術(shù)研究已成為石油加工領(lǐng)域非常活躍的研究課題。
當(dāng)前,吸附法[3-4]、氧化法[5-6]以及生物化學(xué)法[7-8]等非加氫脫硫技術(shù)得到廣泛研究,以期實(shí)現(xiàn)工業(yè)化程度的深度脫硫。其中,以綠色氧化劑過(guò)氧化氫為氧源、雜多酸及其鹽為催化劑的催化氧化脫硫體系被廣泛認(rèn)為是一種最具優(yōu)勢(shì)的超深度催化脫硫技術(shù)。燃油中的硫化物(噻吩、二苯并噻吩及其衍生物等)在該體系中被選擇性地氧化為砜,然后利用液-液萃取手段將極性較強(qiáng)的砜除去,具有操作簡(jiǎn)便、催化活性高、無(wú)二次污染等優(yōu)勢(shì)。如Lucy等[9]設(shè)計(jì)了一種磷鎢酸-過(guò)氧化氫體系,對(duì)二苯并噻吩進(jìn)行了催化氧化研究。結(jié)果表明,最佳條件下目標(biāo)分子轉(zhuǎn)化率達(dá)到了100%。將此項(xiàng)研究應(yīng)用到汽油脫硫工藝中,可以使硫化物全部氧化。但研究發(fā)現(xiàn),過(guò)氧化氫的分解與二苯并噻吩的轉(zhuǎn)化之間存在競(jìng)爭(zhēng),導(dǎo)致該工藝在實(shí)現(xiàn)工業(yè)化應(yīng)用方面還略有不足:①氧化劑使用效率較低;②催化劑回收再利用困難;③硫化物氧化選擇性較差;④易引入其他成分。我國(guó)科學(xué)家在深度催化氧化脫硫方面也做了大量工作,如Meng等[10]成功合成了一系列磷酸咪唑鹽型離子液體,以其為溶劑,使用七鉬酸銨四水合物為催化劑,過(guò)氧化氫為氧化劑,對(duì)4,6-二甲基二苯并噻吩進(jìn)行了催化氧化。結(jié)果表明,該體系表現(xiàn)出了非常高的催化性能,在50℃反應(yīng)3h,4,6-二甲基二苯并噻吩的轉(zhuǎn)化率即可達(dá)到89.2%。另外,該體系反應(yīng)條件溫和,不影響燃料油品質(zhì),體系可以重復(fù)利用6次以上。
上述研究體系可以有效達(dá)到深度脫硫的要求,但在實(shí)現(xiàn)工業(yè)應(yīng)用方面仍存在一定的限制,主要問(wèn)題是反應(yīng)后催化劑很難從體系中分離,一方面增加了催化劑的成本;另一方面造成了二次污染。而通過(guò)使用擔(dān)載型催化劑,可有效解決這一難題,在已報(bào)道的研究工作中,擔(dān)載型催化劑在催化氧化反應(yīng)中已顯示出明顯的優(yōu)勢(shì)[11-12]。本文作者在正庚烷溶劑中,以PNIPAM高聚物微凝膠為模板,通過(guò)外源沉積技術(shù)制備了表面負(fù)載過(guò)氧磷鎢雜多酸十六烷基季胺鹽的雜化微球。利用SEM、FT-IR和TGA等技術(shù)對(duì)所得雜化微球進(jìn)行了組成與形貌表征。然后,以合成的雜化微球?yàn)榇呋瘎?0% H2O2為氧化劑,在十氫化萘、四氫化萘和正戊烷混合溶劑中,對(duì)硫化物噻吩、DBT以及4,6-二甲基二苯并噻吩進(jìn)行了模擬催化氧化研究。結(jié)果表明,雜化微球?qū)︵绶浴BT以及4,6-二甲基二苯并噻吩均具有較高的催化性能。其中,在最佳條件下,小分子噻吩可短時(shí)間內(nèi)被完全氧化,DBT最大轉(zhuǎn)化率達(dá)到了90%以上。而且,催化劑經(jīng)處理后可循環(huán)利用3次以上。
1 實(shí)驗(yàn)部分
1.1 實(shí)驗(yàn)試劑
正庚烷,Span-80,過(guò)硫酸胺(APS),N,N,N',N'-四甲基乙二胺(TMED),N-異丙基丙烯酰胺(NIPAM),N,N'-亞甲基雙丙稀酰胺(BA),丙酮,磷鎢酸(HPW),十六烷基三甲基溴化胺(CTAB),30%過(guò)氧化氫,噻吩,二苯并噻酚(DBT),4,6-二甲基二苯并噻吩(4,6-DMDBT),乙腈,十氫化萘,四氫化萘和正戊烷,以上試劑均為分析純。實(shí)驗(yàn)所用水均為去離子水。
1.2 實(shí)驗(yàn)方法
(1)PNIPAM微凝膠的制備 如已報(bào)道文獻(xiàn)所述[13],將0.6g Span-80 加入到100mL正庚烷溶劑中,通入氮?dú)膺M(jìn)行保護(hù),保持?jǐn)嚢杷俣?00r/min,20℃下充分乳化30min。稱(chēng)取1.2g NIPAM,1mL BA和0.5mL APS相繼溶于6mL去離子水中,配成水相。將配成的水相加入到乳化液中,保持轉(zhuǎn)速不變,繼續(xù)攪拌10min,然后再移入1mL TMED,持續(xù)反應(yīng)約4~5h。停止反應(yīng),固體產(chǎn)物用水和丙酮交替洗滌3~4次后,自然晾干。
(2)PW-HPW/PNIPAM擔(dān)載型催化劑的制 備 量取80mL 正庚烷于150mL 的三口燒瓶中,然后加入1.5g 表面活性劑Span-80,通入氮?dú)猓瑢⒎磻?yīng)裝置置于冰水浴中,攪拌至Span-80均勻分散。接著,將0.12g用HPW的過(guò)氧化氫溶液充分溶脹過(guò)夜的PNIPAM水凝膠移入瓶中。保持轉(zhuǎn)速500r/min不變,再次攪拌30min后,用恒壓漏斗以1滴/秒的速度加入CTAB的過(guò)氧化氫溶液。滴加完畢,轉(zhuǎn)速不變,升高反應(yīng)溫度至50℃,持續(xù)反應(yīng)4~5h。停止反應(yīng)后,固體產(chǎn)物經(jīng)二次水、丙酮交替洗滌3~4次后,室溫下自然晾干。在相同條件下,不使用PNIPAM為模板,制備PW-HPW催化劑粉末,干燥后備用。
(3)模擬催化脫硫?qū)嶒?yàn) 分別量取20mL十氫萘、15mL四氫萘和15mL正戊烷,依次加入100mL的三口燒瓶中。然后,加入0.702mmol DBT(或同物質(zhì)的量的噻吩或4,6-DMDBT)于瓶中,500r/min攪拌至完全溶解,再加入0.1g用30% H2O2溶脹的催化劑PW-HPW/PNIPAM。一定溫度下,調(diào)整轉(zhuǎn)速至1000r/min,持續(xù)反應(yīng)4~5h。停止反應(yīng),過(guò)濾,使用丙酮和水將催化劑交替洗滌3~4次,室溫下自然晾干備用。濾液取樣進(jìn)行GC-FID檢測(cè)。另外,濾液用水與乙腈的混合溶劑萃取,即可將氧化產(chǎn)物砜除去。
1.3 表征
模板及雜化微球的形貌在PhilipXL-20型掃描電子顯微鏡上觀(guān)察,并利用在線(xiàn)電子能譜進(jìn)行元素分析,加速電壓15kV或20kV。紅外光譜在AVTAR360 Nicolet FTIR 光譜儀上測(cè)定。使用Perkin-Elmer TGA-7型熱重分析儀對(duì)材料熱穩(wěn)定性及表面擔(dān)載物含量進(jìn)行測(cè)定。使用毛細(xì)管柱Agilen 6890N GC-FID氣相色譜儀確定DBT的轉(zhuǎn)化率。
2 結(jié)果與討論
2.1 催化劑表征
對(duì)合成的微凝膠及雜化微球的形貌進(jìn)行了掃描電鏡觀(guān)察,如圖1所示。對(duì)于PNIPAM微凝膠[圖1(a) 、(b)],可以看出,微凝膠呈規(guī)則的球形形貌,顆粒直徑約70~90μm,表面光滑致密。在實(shí)驗(yàn)中發(fā)現(xiàn),該微凝膠顆粒具有可逆的溶脹收縮過(guò)程。與PNIPAM模板相比,本實(shí)驗(yàn)所合成的表面沉積有雜多酸季銨鹽的雜化微球也具有球形形貌[圖1(c) 、(d)],單分散性較好,顆粒直徑大約為150~200μm。不同的是,復(fù)合微球表面具有由小褶皺構(gòu)成的規(guī)則形貌。這種微凝膠表面圖案化的成因,已被大量實(shí)驗(yàn)所證實(shí),來(lái)自于由模板的“限域沉積”現(xiàn)象,在胡道道小組[14-16]的相關(guān)報(bào)道中已被指出。
使用紅外光譜對(duì)實(shí)驗(yàn)產(chǎn)物的組成進(jìn)行了表征,結(jié)果如圖2所示。在PNIPAM微凝膠紅外譜圖中,很明顯,1665cm-1處的強(qiáng)吸收峰為C=O伸縮振動(dòng)峰,胺基的伸縮振動(dòng)峰位于3500~3300cm-1處[17-18]。與PNIPAM相比,PW-HPW/PNIPAM雜化微球的紅外譜圖中出現(xiàn)了明顯的過(guò)氧磷鎢酸季胺 鹽的特征峰,875cm-1左右峰是(O—O)的振動(dòng)峰,969cm-1出現(xiàn)的峰是(W=O)的振動(dòng)峰[19]。根據(jù)文獻(xiàn)報(bào)道,在540cm-1和604cm-1左右存在(O—W—O)的振動(dòng)吸收峰[20],但本研究中,由于載體PNIPAM在該區(qū)域存在較強(qiáng)的吸收帶,導(dǎo)致這兩個(gè)峰表現(xiàn)得并不十分明顯。

圖1 PNIPAM微凝膠和PW-HPW/PNIPAM雜化微球的掃描電鏡照片
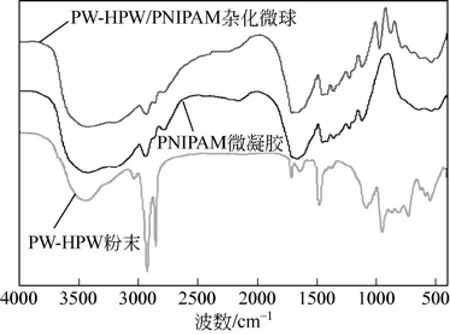
圖2 PW-HPW粉末、PNIPAM微凝膠和PW-HPW/PNIPAM雜化微球的紅外光譜圖
與紅外表征結(jié)果一致,雜化微球表面的能譜分析也確證了雜多酸季銨鹽被成功沉積于PNIPAM高聚物微凝膠表面,如圖3所示。圖中元素Au來(lái)自電鏡觀(guān)察前樣品表面所鍍的金膜;元素C、N和O主要為載體PNIPAM的組成元素,另外一部分C、N和O來(lái)自胺鹽中的CTAB;而元素P和W則是PW-HPW特有的元素。此外,考慮到催化劑的熱穩(wěn)定性對(duì)其在工業(yè)應(yīng)用方面具有非常重要的影響,使用熱重分析儀對(duì)雜化微球的熱分解行為進(jìn)行了研究,如圖4所示。雜化微球受熱失重行為主要分為兩個(gè)階段,200℃前主要失去的是吸附水和溶劑,雜化微球從310℃開(kāi)始受熱分解,直至600℃分解完成,有機(jī)物被完全除去,殘留物為WO3和P2O5。上述熱分解行為表明雜化微球具有較高的熱穩(wěn)定性,可以承受300℃以下高溫。
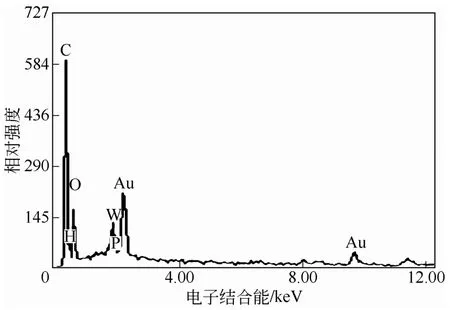
圖3 表面能譜分析
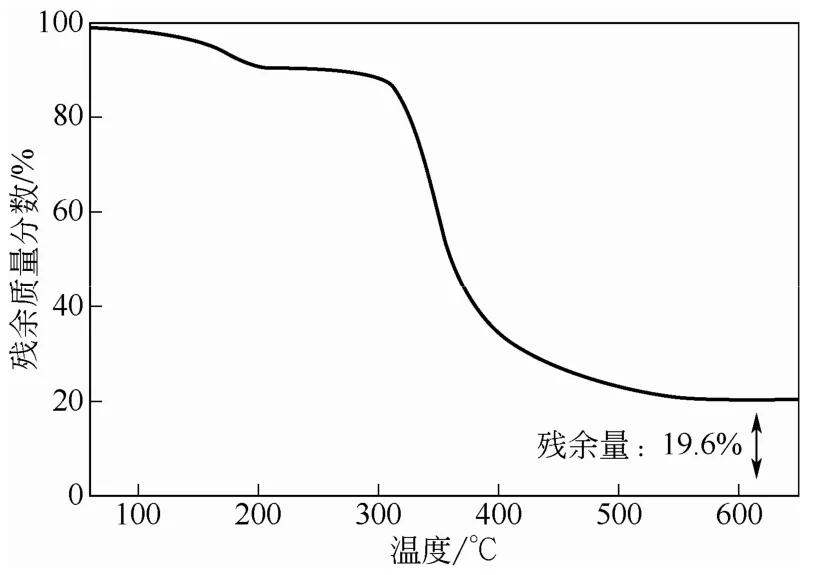
圖4 熱重分析曲線(xiàn)
2.2 催化劑催化性能
2.2.1 反應(yīng)溫度和時(shí)間的影響
以合成的擔(dān)載型雜化微球?yàn)榇呋瘎?0% H2O2為氧化劑,其他條件與1.2節(jié)相同,分別在20℃、30℃、40℃和60℃下進(jìn)行DBT催化氧化實(shí)驗(yàn),通過(guò)不同時(shí)間取樣測(cè)定DBT轉(zhuǎn)化率,考察了溫度和時(shí)間對(duì)催化劑催化效率的影響,如圖5所示。可以看出,40℃之前,隨著溫度的升高,DBT轉(zhuǎn)化率也隨之升高。但隨著進(jìn)一步升高溫度,60℃時(shí)催化劑效率略有降低。根據(jù)已有相關(guān)報(bào)道結(jié)果,升高溫度在提高了催化劑催化效率的同時(shí),也加速了氧化劑H2O2的分解,這應(yīng)該是造成高溫下DBT轉(zhuǎn)化率下降的主要原因[21]。圖5同時(shí)給出了反應(yīng)時(shí)間對(duì)DBT催化氧化率的影響。曲線(xiàn)變化趨勢(shì)表明反應(yīng)初期DBT轉(zhuǎn)化較快,反應(yīng)后期,隨著氧化劑被逐漸消耗,轉(zhuǎn)化速度下降,反應(yīng)8h后,催化反應(yīng)基本完成,40℃時(shí)DBT最大轉(zhuǎn)化率可達(dá)90%以上。
2.2.2 氧化劑用量的影響
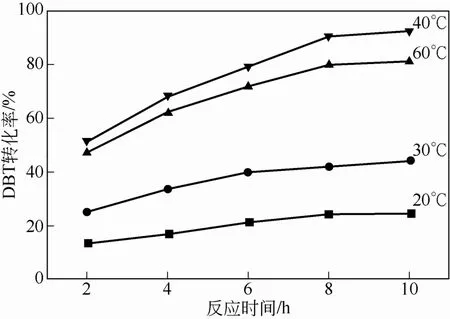
圖5 分別在20℃、30℃、40℃和60℃下DBT的轉(zhuǎn)化率曲線(xiàn)
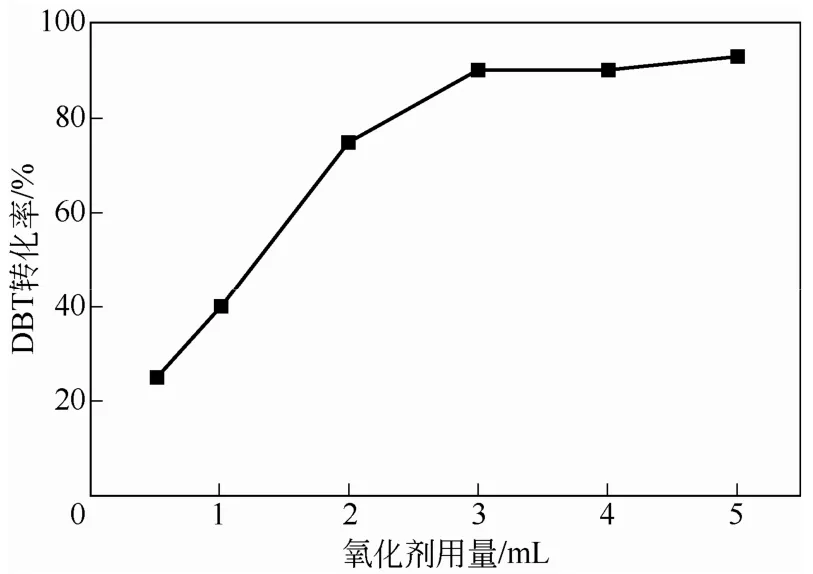
圖6 氧化劑用量對(duì)DBT轉(zhuǎn)化率的影響
通過(guò)使用不同體積的30% H2O2溶脹PW-HPW/PNIPAM雜化微球,在其他實(shí)驗(yàn)條件與1.2 節(jié)相同的情況下,對(duì)DBT進(jìn)行了催化氧化實(shí)驗(yàn),考察了氧化劑用量對(duì)DBT轉(zhuǎn)化率的影響,如圖6 所示。圖中曲線(xiàn)變化表明,隨氧化劑用量的增加,DBT轉(zhuǎn)化率明顯增加。過(guò)氧化氫用量為3mL時(shí),轉(zhuǎn)化率為90.32%。繼續(xù)增加氧化劑用量,轉(zhuǎn)化率略有增加,但增幅不大。實(shí)驗(yàn)中發(fā)現(xiàn),雜化微球?qū)^(guò)氧化氫的吸附存在極限,當(dāng)氧化劑用量為3mL時(shí),吸附飽和,此時(shí)增加氧化劑用量,過(guò)多的氧化劑將分散于反應(yīng)體系,無(wú)法被雜化微球吸附。因此,氧化劑的用量以3mL為佳。
2.2.3 與非擔(dān)載催化劑催化效率的對(duì)比
為了考察擔(dān)載行為對(duì)催化劑催化性能的影響,保持其他反應(yīng)條件不變,在40℃下,使用30% H2O2為氧化劑,DBT為硫化物,分別進(jìn)行了無(wú)催化劑的空白實(shí)驗(yàn)和過(guò)氧磷鎢雜多酸季胺鹽粉末為催化劑的對(duì)比實(shí)驗(yàn)。同時(shí),分別考察了雜化微球?qū)α蚧镟绶院?,6-DMDBT的催化氧化性能,如圖7所示。從圖7中曲線(xiàn)變化趨勢(shì)可以看出,空白試驗(yàn)條件下,DBT轉(zhuǎn)化率幾乎為零;非擔(dān)載型催化劑催化效率最高,反應(yīng)8h DBT幾乎完全轉(zhuǎn)化,表明固載后的雜化微球催化效率略低。但需要指出的是,非擔(dān)載型催化劑反應(yīng)后很難從體系回收,而雜化微球可以通 過(guò)離心從體系中分離,經(jīng)處理后重復(fù)使用。另外,在對(duì)噻吩和4,6-DMDBT的催化氧化實(shí)驗(yàn)中,4,6-DMDBT轉(zhuǎn)化能力略低于DBT,而噻吩6h后即可達(dá)到完全轉(zhuǎn)化,表明小分子硫化物更易被催化氧化,與相關(guān)研究報(bào)道結(jié)果一致[22]。上述實(shí)驗(yàn)結(jié)果表明,表面擔(dān)載過(guò)氧雜多酸的雜化微球?qū)︵绶浴BT及4,6-DMDBT均具有較強(qiáng)的催化性能。
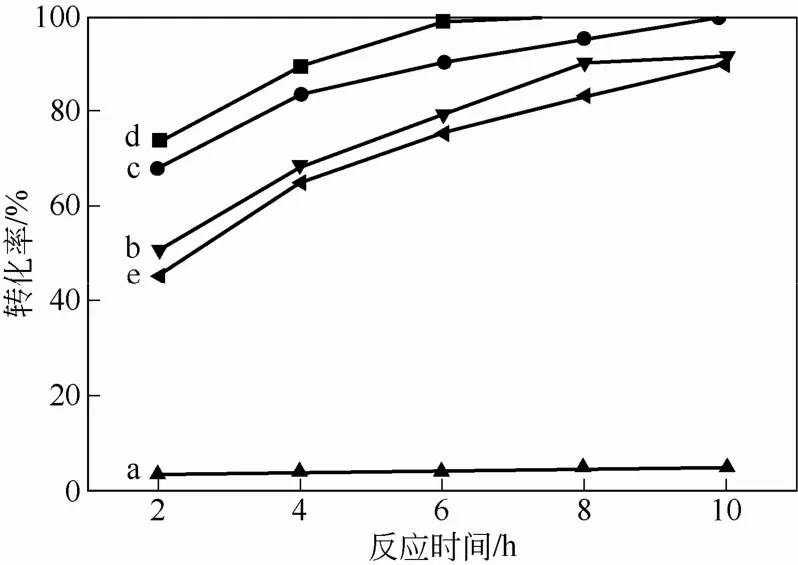
圖7 雜化微球?qū)α蚧镟绶院?,6-DMDBT的催化氧化性能
2.2.4 催化劑重復(fù)利用效率
反應(yīng)結(jié)束后,通過(guò)過(guò)濾將復(fù)合微球催化劑從體系中分離出來(lái),用丙酮和水交替洗滌處理后,室溫下干燥。接著,處理后的雜化微球用30% H2O2重新溶脹,再次投入循環(huán)使用。在40℃下,以DBT為硫化物,考察了催化劑重復(fù)使用效率,結(jié)果見(jiàn)圖8。可以明顯看出,重復(fù)使用3次,催化劑活性基本 保持不變。隨著重復(fù)次數(shù)增加,催化劑活性開(kāi)始下降,應(yīng)該是雜化微球表面活性組分脫落造成的。
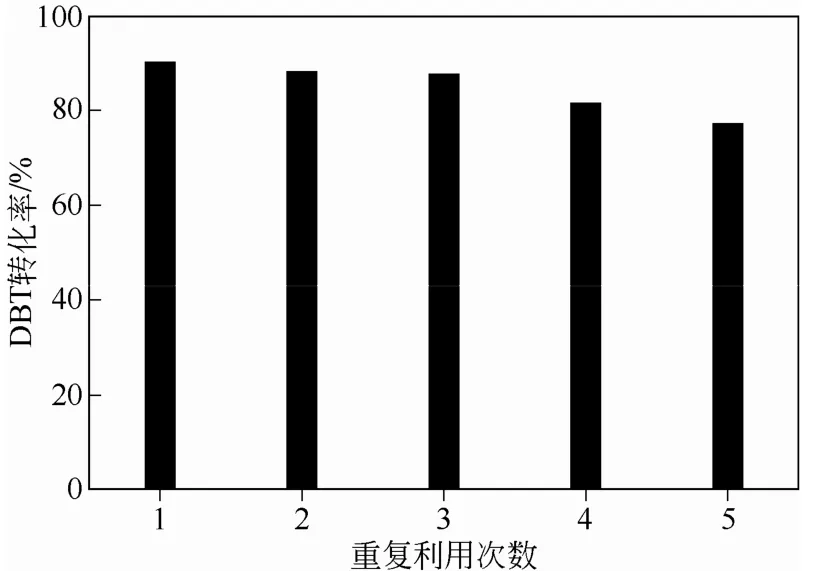
圖8 催化劑重復(fù)實(shí)驗(yàn)效率
2.2.5 催化氧化脫硫機(jī)理
基于上述實(shí)驗(yàn)表征結(jié)果,對(duì)PW-HPW/PNIPAM雜化微球作用下、以30% H2O2為氧化劑時(shí)硫化物DBT的催化氧化機(jī)理進(jìn)行了探討,如圖9所示。反應(yīng)起始,在攪拌作用下,雜化微球被均勻分散于反應(yīng)體系,在雜化微球表面,沉積的雜多酸季胺鹽作為相轉(zhuǎn)移催化劑,使得微球內(nèi)部的H2O2與DBT接觸,并繼而將DBT氧化為砜。催化氧化反應(yīng)結(jié)束,催化氧化活性成分由過(guò)氧磷鎢酸季銨鹽變?yōu)榉腔钚粤祖u酸季胺鹽。在雜化微球內(nèi)部的H2O2作用下,非活性磷鎢酸季胺鹽重新被原位氧化為過(guò)氧磷鎢酸季胺鹽催化劑。催化氧化反應(yīng)循環(huán)反復(fù)進(jìn)行,直至H2O2消耗完全為止。反應(yīng)結(jié)束后,停止攪拌,雜化微球沉積于反應(yīng)器底部。利用過(guò)濾,將雜化微球分離出來(lái),活化后可重復(fù)利用。
3 結(jié) 論

圖9 PW-HPW/PNIPAM雜化微球作用下DBT催化氧化機(jī)理示意圖
以PNIPAM高聚物微凝膠為模板,成功合成了表面擔(dān)載有過(guò)氧磷鎢雜多酸十六烷基季胺鹽的雜化微球,并將其作為催化劑用于模擬催化氧化脫硫?qū)嶒?yàn)。結(jié)果表明,使用過(guò)氧化氫為氧化劑,雜化微球不僅對(duì)二苯并噻吩具有較高的催化效率,而且對(duì)噻吩與4,6-二甲基二苯并噻吩也表現(xiàn)出較強(qiáng)的催化活性。尤為重要的是,該體系為非均相催化氧化體系,雜化微球催化劑能夠從體系中進(jìn)行簡(jiǎn)易分離,經(jīng)活化處理后可多次重復(fù)利用,而催化性能下降不大。本文僅對(duì)雜化微球的催化氧化脫硫性能進(jìn)行了初步的模擬研究,有關(guān)雜化微球在催化深度脫硫方面應(yīng)用研究的許多細(xì)節(jié)性工作正在進(jìn)行之中。但目前實(shí)驗(yàn)結(jié)果已表明,擔(dān)載型雜化微球在深度脫硫方面具有獨(dú)特的優(yōu)勢(shì),這種非均相催化氧化體系不僅有望實(shí)現(xiàn)綠色催化氧化深度脫硫,并且對(duì)構(gòu)筑非均相催化微反應(yīng)器具有普遍的借鑒意義。
[1] US EPA. Regulatory announcement:Heavy-duty engine and vehicle standards and highway diesel fuel sulfur control requirements. EPA420-F-00-057[EB/OL]. 2000. http://www.epa.gov/otaq/diesel. htm.
[2] 李宇慧,馮麗娟,王景剛,等. MoO3/Al2O3-H2O2體系用于柴油催化氧化脫硫[J]. 化工進(jìn)展,2010,29(s1):659-661.
[3] Yang R T,Hernandez-Maldonado A J,Yang F H. Desulfurization of transportation fuels with zeolites under ambient conditions[J].Science,2003,301:79-81.
[4] Gao X,Du Z,Ding H,et al. Kinetics of NOxabsorption into (NH4)2SO3solution in an ammonia-based wet flue gas desulfurization process[J].Energy Fuels,2010,24(11):5876-5882.
[5] Chapados D,Bonde S E,Gore W L,et al. NPRA AM-00-25[R]. Washington D C:National Petrochemical and Refiners Association,2008.
[6] Filippis P D,Liuzzo G,Scarsella M,et al. Oxidative desulfurization Ⅱ:Temperature dependence of organosulfur compounds oxidation[J].Ind. Eng. Chem. Res.,2011,50(18):10452-10457.
[7] Boron D J,Deever W R,Atlas R M,et al. NPRA AM-99-54[R]. Washington D C:National Petrochemical and Refiners Association,1999.
[8] Vazquez-Duhalt R,Torres E,Valderrama B,et al. Will biochemical catalysis impact the petroleum refining industry?[J].Energy Fuels,2002,16(5):1239-1250.
[9] Collins F M,Lucy A R,Sharp C,et al. Oxidative desulphurisation of oilsviahydrogen peroxide and heteropolyanion catalysis[J].J. Mol. Catal. A:Chem.,1997,117:397-403.
[10] Shao B,Shi L,Meng X. Deep desulfurization of 4,6-dimethyldienzothiophene by an ionic liquids extraction coupled with catalytic oxidation with a molybdic compound[J].Ind. Eng. Chem. Res.,2014,53:6655-6663.
[11] 董陽(yáng)陽(yáng),白金泉,王喬喬. 介孔分子篩固載希夫堿金屬配合物催化氧化研究進(jìn)展[J]. 化學(xué)試劑,2013,35(3):233-238.
[12] 姜偉麗,豆丙乾,李沛東,等. 烯烴歧化反應(yīng)中的負(fù)載氧化鎢催化劑[J]. 化工進(jìn)展,2012,31(12):2686-2719.
[13] 宋少飛,周福林,弓巧娟. 氣相原位反應(yīng)制備表面負(fù)載納米銀PAM復(fù)合微球[J]. 陜西師范大學(xué)學(xué)報(bào):自然科學(xué)版,2013,41(3):36-39.
[14] Yang J X,F(xiàn)ang Y,Bai C L,et al. CuS-poly (N-isopropylacrylamide-co-acrylic acid) composite microspheres with patterned surface structures:Preparation and characterization[J].Chin. Sci. Bull.,2004,49:2026-2032.
[15] Yang J X,Hu D D,F(xiàn)ang Y,et al. Novel method for preparation of structural microspheres poly(n-isopropylacrylamide-co-acrylic acid)/SiO2[J].Chem. Mater.,2006,18:4902-4907.
[16] Wang X J,Hu D D,Yang J X. Synthesis of PAM/TiO2composite microspheres with hierarchical surface morphologies[J].Chem. Mater.,2007,19:2610-2621.
[17] 周美娟,魏長(zhǎng)平,畢穎麗,等. 磷鎢雜多化合物催化H2O2氧化十八醇制十八酸[J]. 催化學(xué)報(bào),1999,20(4):437-441.
[18] 張乾,范曉東. 丙烯酞胺反相微乳液體系的制備、聚合及表征[J].化學(xué)工業(yè)與工程,2001,18(6):316-340.
[19] 陳小華,陳傳盛,孫磊,等. 碳納米管的表面修飾及其在水中的分散性能研究[J]. 湖南大學(xué)學(xué)報(bào),2004,31(5):18-21.
[20] Sun Y,Xi Z,Cao G Y. Epoxidation of olefins catalyzed by[π-C5H5NC16H33]3[PW4O16] with molecular oxygen and a recyclable reductant 2-ethylanthrahydroquinone[J].J. Mol. Catal. A:Chemical,2001,166:219-224.
[21] 田雪,劉淑芝,劉英楠,等. 過(guò)氧磷鎢酸催化氧化柴油深度脫硫?qū)嶒?yàn)研究[J]. 化工科技,2012,20(2):17-20.
[22] 宋華,穆金城. 催化氧化脫硫分子篩催化劑研究進(jìn)展[J]. 化工進(jìn)展,2011,30(2):303-308.
猜你喜歡
小獼猴智力畫(huà)刊(2022年9期)2022-11-04 02:31:42
中學(xué)生數(shù)理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學(xué))(2019年6期)2019-10-10 01:01:50
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
發(fā)明與創(chuàng)新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
浙江大學(xué)學(xué)報(bào)(工學(xué)版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國(guó)資源綜合利用(2016年4期)2016-01-22 08:27:23
