小分子抗腫瘤藥物SPH0396在大鼠體內的藥代動力學
2015-09-14 08:10:08馬晨向志雄萬惠新劉基曄夏廣新上海醫藥集團股份有限公司中央研究院上海201203
上海醫藥 2015年11期
關鍵詞:血漿
馬晨 向志雄 萬惠新 劉基曄 夏廣新(上海醫藥集團股份有限公司中央研究院 上海 201203)
小分子抗腫瘤藥物SPH0396在大鼠體內的藥代動力學
馬晨 向志雄 萬惠新 劉基曄 夏廣新
(上海醫藥集團股份有限公司中央研究院 上海 201203)
目的:建立定量測定大鼠血漿中抗腫瘤藥aSPH0396的LC-MS/MS方法,并將其應用于大鼠體內藥代動力學研究。方法:血漿樣品在Waters BEH C18柱上以0.1%甲酸水溶液-0.1%甲酸乙腈溶液進行梯度洗脫;采用MRM方式進行定量測定,監測離子對為m/z 553.4→453.4(SPH0396)和m/z 533.3→259.9(內標ponatinib)。結果:SPH0396在1.11~2 488.50 ng/ml的濃度范圍內呈良好的線性關系(R2=0.999),定量下限為1.11 ng/ml,回收率和精密度均符合生物樣品檢測要求。大鼠靜注3 mg/kg SPH0396后,t1/2為3.43±0.37 h,CL為2.13±0.21L/h·kg,Vdss為6.76±0.26 L/kg。大鼠口服5、15 和50 mg/kg的SPH0396后吸收較慢,Tmax為4~6 h。大鼠口服不同劑量的SPH0396,Cmax和AUC(0-t)的增加比例均高于劑量增加比例,生物利用度分別為17.15%、25.58%和36.19%。結論:本研究首次建立了特異、靈敏、便捷的定量檢測大鼠血漿中SPH0396的LC-MS/MS方法,并成功應用于SPH0396的大鼠藥代動力學研究。
酪氨酸激酶抑制劑 藥代動力學 LC-MS/MS
酪氨酸激酶(TKs)在許多生理過程都發揮關鍵性作用,而它的活性失調被證明與多種癌癥的發生、發展相關。近年來,以酪氨酸激酶為靶點進行藥物研發已成為國際上抗腫瘤藥物研究的熱點,多種小分子酪氨酸激酶抑制劑(TKIs)被批準上市用于腫瘤治療[1-3]。本研究院針對Bcr-Abl靶點[4-7],設計并合成了一系列化合物,并對其中部分化合物進行了體外藥效學和早期代謝性質的評價。其中SPH0396(圖1)在激酶和細胞水平的抗腫瘤活性均接近或優于上市藥物bosutinib[8-11](圖1),體內外藥代性質良好。因此將其選為候選化合物進一步開發,用于對伊馬替尼耐藥的慢性粒細胞白血病(CML)。本文對SPH0396在雄性大鼠體內的藥代動力學性質進行了研究。
1 材料與儀器
1.1 藥品與試劑
SPH0396由上海醫藥中央研究院提供,純度99.69%;內標帕納替尼(ponatinib,圖1;Molttl公司);色譜純級乙腈(德國Merck公司);色譜純級甲酸(德國CNW公司);其它分析純級試劑均購自中國醫藥(集團)上海化學試劑公司;實驗用純水由Milli-Q純水儀制備而成。
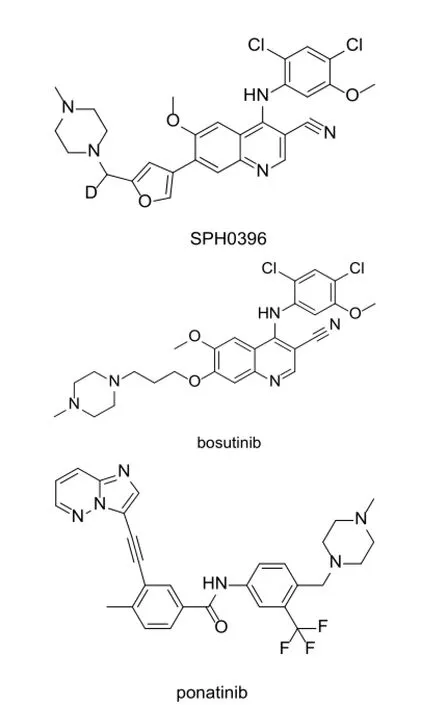
圖1 SPH0396, bosutinib 和ponatinib的化學結構
1.2 儀器
Acquity UPLC液相色譜系統(美國Waters公司)串聯API4000 Q-trap型三重四級桿質譜檢測器(美國Applied Biosystems公司),配有ESI源、四元梯度泵、自動進樣器和數據處理系統;CPA225D分析天平(德國Satroious公司);Mulrifuge X3R高速冷凍離心機(美國Thermo公司);V230多管振蕩器(美國Fisher公司);Milli-QTM型超純水凈化器(美國Millipore公司)。
1.3 實驗動物
SD大鼠,雄性,180~220 g,由上海西普爾必凱實驗動物有限公司提供。許可證號SCXK(滬)2013~0016。
2 實驗方法
2.1 LC/MS檢測條件
色譜條件:色譜柱為BEH C18柱(2.1 mm×50 mm,1.7 mm,美國 Waters公司)。流動相A為0.1%甲酸水溶液,流動相B為0.1%甲酸乙腈溶液,洗脫程序如下:0~0.5 min 流動相A和B的比例為95∶5,0.5~1.0 min線性變至5∶95,1.0~2.2 min維持5∶95,2.2~2.5 min線性恢復至95∶5,2.5~3 min保持95∶5。流速0.3 ml/min,柱溫40 ℃,進樣量5 ml。
質譜條件:離子源為ESI源,正離子模式檢測;毛細管電壓5 500 V,離子源溫度500 ℃,干燥氣10 L/ min,霧化氣50 psi,碰撞氣壓力(CAD)為Medium;掃描方式為多反應監測(MRM),用于定量分析的離子對分別為SPH0396,m/z 553.4→453.4,去簇電壓(DP)80 V,碰撞能量(CE)30 V;內標ponatinib,m/z 533.3→259.9,DP143 V,CE 30 V。
2.2 樣品處理方法
大鼠血漿樣品50 ml,加入200 ml含內標(ponatinib,0.25 mmol/L)的乙腈沉淀蛋白,渦旋振蕩10 min后,于6 000 g離心10 min,取200 ml上清于6 000 g再次離心10 min,取上清50 ml于96孔板中進樣,進樣量5 ml。
2.3 LC/MS方法學驗證
2.3.1 標準曲線的配制
精密稱取SPH0396粉末5.03 mg,加入90.9 ml的DMSO,配制為100 mmol/L的儲備液。再將此儲備液用乙腈逐級稀釋成濃度為22.14、55.35、166.04、553.46、1 600.38、5 534.60、16 603.80、33 207.60、49 811.40 ng/ ml的系列工作液。取95 ml的空白大鼠血漿加入5 ml的上述工作液,配制成濃度為1.11、2.77、8.30、27.67、83.02、276.73、830.19、1 600.38、2 490.57 ng/ml的標準曲線。取50 ml的含藥血漿按2.2項下方法處理后進樣。
2.3.2 內標液的配制
精密稱取ponatinib 2.53 mg,加入47.5 ml的DMSO,配制為100 mmol/L的儲備液。取上述儲備液2.5 ml,加入1 L乙腈,配制為0.25 mmol/L的內標液。
2.3.3 質控樣品的配制
將2.3.1中100 mmol/L的SPH0396儲備液用乙腈逐級稀釋成濃度為49.81、11 069.20和38 742.20 ng/ml的系列質控工作液。取95 ml的空白大鼠血漿加入5 ml的質控工作液,配制成濃度為2.49、553.46、1 937.11 ng/ ml的質控樣品。取50 ml的含藥血漿按2.2項下方法處理后進樣。
2.4 大鼠體內實驗
SD大鼠16只,雄性,180~220 g。分為4組,每組4只。分別靜脈注射3 mg/kg或灌胃給予5、15和50 mg/kg的SPH0396,于給藥前和給藥后2、5、15、30、60、90、120、240、360、480、600、1 440 min分別于大鼠眼底靜脈叢取血0.2 ml。血樣于8 000 r/min離心5 min,取血漿于離心管中-20 ℃保存待測。
2.5 數據處理方法
SPH0396的血藥濃度-時間曲線用Kinetica軟件(Version 5.1,美國)的非房室模型進行擬合,得到藥代動力學參數。Cmax為實測的最大血藥濃度,Tmax為口服給藥后血藥濃度達峰時間,t1/2為末端消除半衰期,MRT為體內平均滯留時間,血藥濃度-時間曲線下面積AUC(0-t)由梯形面積法計算得到,Vdss為穩態時表觀分布容積,CL為清除率。絕對生物利用度計算公式如下:
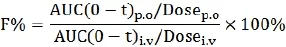
注:i.v為靜脈注射;p.o為口服
3 實驗結果
3.1 方法的專屬性
用來自6只大鼠的空白血漿配制標準樣品,空白血樣、空白血樣加標準品和大鼠給藥后4 h血樣的MRM色譜圖見圖2。大鼠血漿中的內源性物質不干擾SPH0396和內標ponatinib的測定,SPH0396的保留時間為1.59 min,ponatinib的保留時間為1.95 min,表明方法專屬性良好。
3.2 標準曲線與線性范圍
按2.3.1項下方法配制標準曲線樣品并進行測定,以SPH0396樣品峰面積比ponatinib內標峰面積(As/Ai)對濃度(C)作圖,得到標準曲線方程為y=0.265 7x+0.007 6,相關系數R2=0.999。線性范圍為1.11~2 490.57 ng/ml,定量下限為1.11 ng/ml。
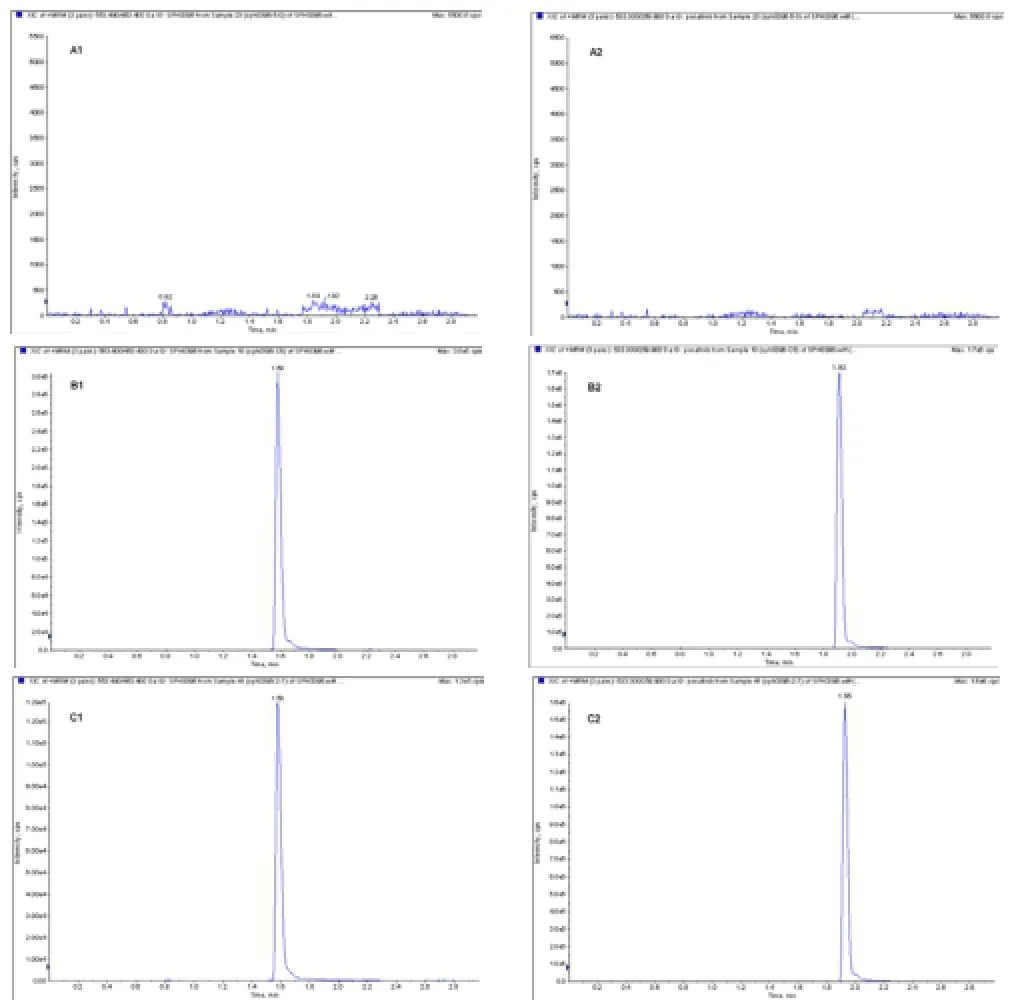
圖2 大鼠血漿中的SPH0396的MRM色譜圖
3.3 精密度和準確度
配制低、中、高3個濃度(2.49、553.46、1 937.11 ng/ml)的SPH0396質控樣品各6份,用隨行標準曲線計算質控樣品測得濃度。以質控樣品測得濃度與加樣濃度相比計算方法的準確度與精密度(表1)。RSD均小于10%。
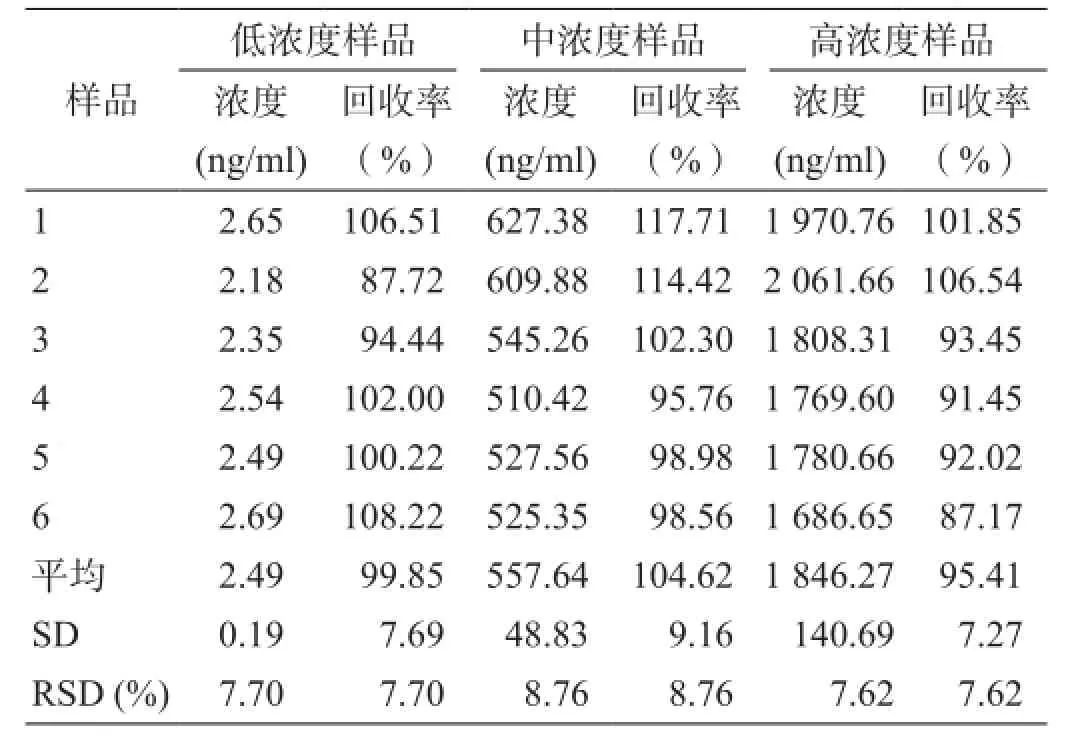
表1 LC-MS/MS測定方法的精密度與準確度
3.4 大鼠靜脈注射SPH0396后的藥代動力學
大鼠單次靜脈注射3 mg/kg的SPH0396后采集血樣,按2.2項下方法處理后進樣,測定SPH0396在大鼠血漿中的濃度,采用Kinetica計算藥動學參數(表2)。大鼠靜脈注射SPH0396后,AUC(0-t)為1 391.08±132.92 h·ng/ml,消除半衰期為3.43±0.37 h,清除率為2.13±0.21 L/h·kg,表觀分布容積為6.76±0.26 L/kg。
表2 大鼠靜脈注射或口服SPH0396后的藥代動力學參數(±s,n=4)
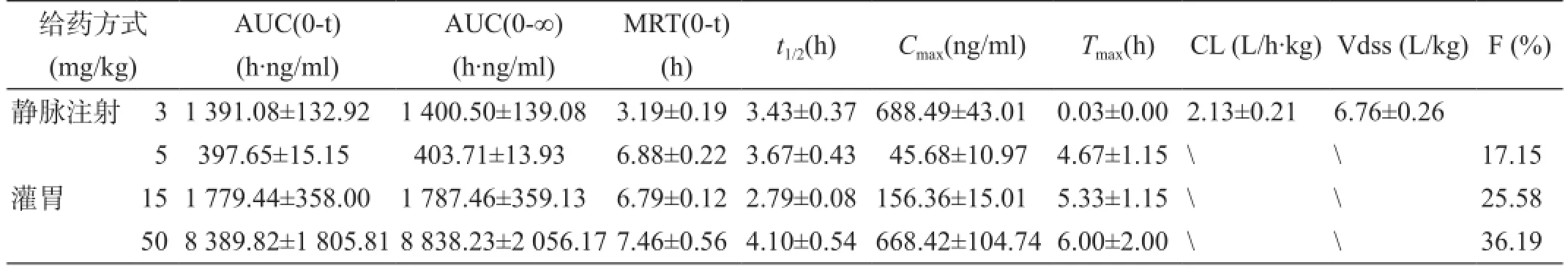
表2 大鼠靜脈注射或口服SPH0396后的藥代動力學參數(±s,n=4)
給藥方式(mg/kg) AUC(0-t) (h·ng/ml) AUC(0-∞) (h·ng/ml) MRT(0-t) (h)t1/2(h)Cmax(ng/ml)Tmax(h)CL (L/h·kg)Vdss (L/kg)F (%)靜脈注射31 391.08±132.921 400.50±139.083.19±0.193.43±0.37688.49±43.010.03±0.002.13±0.216.76±0.26灌胃5397.65±15.15403.71±13.936.88±0.223.67±0.4345.68±10.974.67±1.15\17.15 151 779.44±358.001 787.46±359.136.79±0.122.79±0.08156.36±15.015.33±1.15\25.58 508 389.82±1 805.818 838.23±2 056.177.46±0.564.10±0.54668.42±104.746.00±2.00\36.19
3.5 大鼠灌胃給予SPH0396后的藥代動力學
大鼠單次口服5、15和50 mg/kg的SPH0396后的血藥濃度-時間曲線見圖3。口服SPH0396在大鼠體內吸收較慢,血藥濃度在4~6 h才能達到峰值;Cmax分別為45.68±10.97、156.36±15.01和668.42±104.72 ng/ ml;AUC(0-t)分別為397.65±15.15、1 779.44±358.00和8 389.82±1 805.81 h·ng/ml。大鼠口服不同劑量的SPH0396,Cmax和AUC(0-t)的增加比例均高于劑量增加比例。相應的大鼠口服不同劑量的SPH0396生物利用度分別為17.15%、25.58%和36.19%。
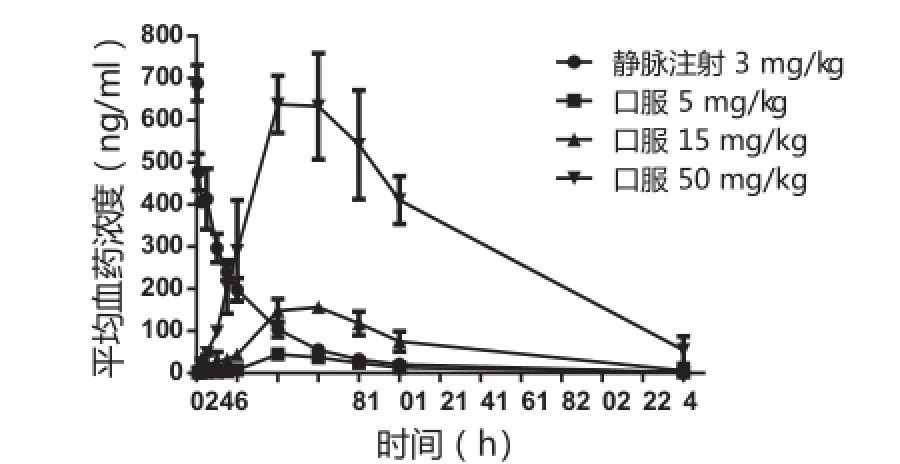
圖3 大鼠靜脈注射或口服SPH0396后的血藥濃度-時間曲線(±s,n=4)
4 討論
SPH0396是一個新結構的抗CML候選化合物。靜脈給藥后,SPH0396在大鼠體內的消除半衰期較長,表觀分布容積大于大鼠總體液量,說明SPH0396在大鼠體內分布較廣。大鼠口服不同劑量的SPH0396,Cmax和AUC(0-t)的增加比例均高于劑量增加比例,表明SPH0396在大鼠體內的吸收是非線性的,提示可能有外排轉運體參與SPH0396的吸收過程,需要進一步實驗加以證實。SPH0396為bosutinib的結構類似物,我們的研究表明,其在大鼠體內的藥動學性質接近或優于bosutinib,在激酶和細胞水平的抗腫瘤活性也均接近或優于bosutinib[9-11],可做為候選化合物進一步開發。
本研究建立并驗證了定量測定大鼠血漿中SPH0396的LC-MS/MS方法。該方法的專屬性好,靈敏度高,回收率和精密度均符合生物樣品的定量分析要求,可用于該化合物在大鼠體內的藥代動力學研究。
[1] Levitzki A. Tyrosine kinase inhibitors: views of selectivity, sensitivity, and clinical performance[J]. Annu Rev Pharmacol Toxicol, 2013, 53: 161-85.
[2] 許嬌紅, 游育紅, 許建華. 酪氨酸激酶抑制劑的研究進展[J]. 海峽藥學, 2010, 22(7): 8-11.
[3] 李欣, 高金恒, 陳國良. 蛋白酪氨酸激酶抑制劑的研究進展[J]. 沈陽藥科大學學報, 2011, 28(12): 1005-1012.
[4] 劉舒暢, 張健存, 陳賽娟. BCR-ABL蛋白激酶抑制劑的研究進展[J]. 中國醫藥工業雜志, 2010, 41(4): 293-297.
[5] Druker BJ, Tamura S, Buchdrunger I, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positivie cells[J]. Nat Med, 1996, 2(5): 561-566.
[6] 王偉, 鄒志紅. BCR-ABL酪氨酸激酶抑制劑的研究進展[J]. 東南大學學報(醫學部), 2010, 29(2): 232-236.
[7] 昌盛, 李曉光. 慢性粒細胞白血病治療藥酪氨酸激酶抑制劑研究進展[J]. 吉林醫藥學院學報, 2012, 33(5): 315-317.
[8] 鄭敏, 白秋江, 胡躍民. 慢性髓性白血病治療藥博舒替尼[J]. 藥物流行病學雜志, 2014, 23(1): 58-60.
[9] Puttini M, Coiluccia AM, Boschelli F, et al. In vitro and in vivo activity of SKI-606, a novel Src-Abl inhibitor, against imatinib-resistant Bcr-Abl+ neoplastic cells[J]. Cancer Res, 2006, 66(23): 11314-11322.
[10] Rabbani SA, Valentino ML, Arakelian A, et al. SKI-606(Bosutinib) blocks prostate cancer invasion, growth, and metastasis in vitro and in vivo through regulation of genes involved in cancer growth and skeletal metastasis[J]. Mol Cancer Ther, 2010, 9(5): 1147-1157.
[11] United States Food and Drug Administration. Pharmacology review approved on 09/04/2012 (PDF) for bosutinib[EB/OL]. (2012-08-15) [2014-12-17]. http: //www.accessdata.fda.gov/ drugsatfda_docs/nda/2012/203341Orig1s000PharmR.pdf.
Pharmacokinetics study of an anti-tumor candidate SPH0396 in rats
MA Chen, XIANG Zhixiong, WAN Huixin, LIU Jiye, XIA Guangxin
(Central Research Institute, Shanghai Pharmaceuticals Holding Co. Ltd., Shanghai 201203, China)
Objective: To develop and validate an LC-MS/MS method for the quantitative analysis of a new antitumor candidate SPH0396 in rat plasma so as to be applied to the pharmacokinetic study in rats. Methods: The chromatographic separation of SPH0396 was performed on BEH C18column by a gradient elution with water containing 0.1% formic acid and acetonitrile containing 0.1% formic acid. The MS detection was conducted on MRM mode and the ion-pairs monitored were m/z 553.4→453.4 (SPH0396) and m/z 533.3→259.9 with ponatinib as an internal standard. Results: A standard curve for the determination of SPH0396 showed a good linearity over the range of 1.11~2 488.50 ng/ml (R2=0.999) with the lower limit of quantification at 1.11 ng/ml. The recovery and precision could meet the requirement of bioanalysis. After intravenous administration of SPH0396, the t1/2, CL and Vdss in rats were 3.43±0.37 h, 2.13±0.21 L/h·kg and 6.76±0.26 L/kg, respectively. The absorption of SPH0396 in rats was slow after oral administration. The peak level was reached at 4~6 h. The oral bioavailability was 17.15%, 25.58% and 36.19% at different doses, respectively. Conclusion: The LC-MS/MS method is specific, sensitive, rapid and simple and is suitable for pharmacokinetic study of SPH0396 in rats.
tyrosine kinase inhibitors; pharmacokinetics; LC-MS/MS
R969.1
A
1006-1533(2015)11-0070-05
2015-01-16)
猜你喜歡
現代臨床醫學(2022年4期)2022-09-29 07:38:00
昆明醫科大學學報(2021年4期)2021-07-23 01:21:50
天津醫科大學學報(2019年6期)2019-08-13 07:04:34
云南醫藥(2019年3期)2019-07-25 07:25:14
現代檢驗醫學雜志(2016年5期)2016-08-20 03:16:56
海南醫學(2016年8期)2016-06-08 05:43:00
西南軍醫(2016年5期)2016-01-23 02:20:33
川北醫學院學報(2015年5期)2015-12-05 08:22:28
醫學研究雜志(2015年9期)2015-07-01 17:28:15
現代檢驗醫學雜志(2015年1期)2015-02-06 01:59:26
