含氟酰氯類化合物的合成研究進展
2015-11-23 07:25:43王術成王軍祥林勝達于萬金劉武燦
浙江化工 2015年2期
關鍵詞:方法
張 迪,王術成,王軍祥,林勝達,于萬金,劉武燦
(1.浙江省化工研究院國家ODS替代品工程技術研究中心,浙江杭州310023;2.中化藍天集團下沙生產基地,浙江杭州310018)
含氟酰氯類化合物的合成研究進展
張 迪1,王術成1,王軍祥2,林勝達1,于萬金1,劉武燦1
(1.浙江省化工研究院國家ODS替代品工程技術研究中心,浙江杭州310023;2.中化藍天集團下沙生產基地,浙江杭州310018)
介紹了包括SO3氧化法、光氧化法、催化氧化法和熱分解法等含氟酰氯類化合物的合成方法,評述了各種合成方法的優缺點,并對含氟酰氯類化合物的合成發展趨勢作出了展望。
氯二氟乙酰氯;二氟乙酰氯;三氟乙酰氯;光氧化;催化氧化
0 前言
含氟酰氯類化合物是一類重要的脂肪族含氟中間體。由于酰氯基團中的氯原子有吸電子效應,并且Cl-也是一個很好的離去基團,因此酰氯具有很強的親核酰基取代反應活性,可以與氨/胺反應生成酰胺(氨解),與醇反應生成酯(醇解),與羧酸根離子反應生成酸酐等,從而向一些物質中引入特定的含氟基團,合成出一系列醫藥和農藥等精細化學品或其中間體。
氯二氟乙酰氯,分子式為C2Cl2F2O,分子量148.92,CAS號354-24-5,沸點20℃,是一種高度揮發性、帶刺激性氣味的無色液體,易水解,在空氣中會發煙,易溶于有機溶劑。氯二氟乙酰氯及其衍生物可用于合成治療瘧疾和癌癥的藥物、氟代氮雜環丁酮類抗生素、吡唑甲酰胺類殺菌劑、除草劑等[1-2]。
二氟乙酰氯,分子式為C2HClF2O,分子量114.48,CAS號381-72-6,沸點25℃,主要用于合成二氟乙酸[3]、二氟乙醇[4]等二氟系列產品及其下游衍生物,如二氟吡唑類殺菌劑、二氟代酮類抑制劑[5]。
三氟乙酰氯,分子式為C2ClF3O,分子量132.47,CAS號354-32-5,熔點-146℃,沸點-27℃,是一種有刺激性氣味的無色氣體,在空氣中易水解、易發煙,易溶于有機溶劑。三氟乙酰氯主要用來合成三氟乙酸或其它衍生物,是合成農藥和醫藥的一種重要原料。
1 合成技術進展
含氟酰氯類化合物最早是通過三氯甲苯/氯化鋅[6]、苯甲酰氯[7-8]、五氯化磷[9]等與相應的含氟羧酸進行反應制得的。該類方法存在的問題是原料含氟羧酸較難取得。目前,根據反應的特點,可以將含氟酰氯類化合物的合成方法分為SO3氧化法、光氧化法、催化氧化法和熱分解法四大類。
1.1 SO3氧化法
1.1.1 以1,1,1,2-四氯-2,2-二氟乙烷為原料制備氯二氟乙酰氯
Haloearbon Prod.專利[10]報道了1,1,1,2-四氯-2,2-二氟乙烷(R112a,CClF2-CCl3)與SO3反應制備一氯二氟乙酰氯的方法。在反應器中加入712 g的Rl12a,3g的HgSO4,3g的Hg2SO4,加熱回流,6 h內加入753 g 65%的發煙硫酸,之后繼續反應1 h,氯二氟乙酰氯的收率為94%。
Kali-Chemie AG專利[11]同樣介紹了通過R112a與SO3反應制備氯二氟乙酰氯的方法,所不同的是以SO2Cl2作為R112a的溶劑。例如,在含有5 mol的SO3、0.4 mol的SO2Cl2、0.25 mol的H2SO4和1 g HgSO4、1 g Hg2SO4催化劑的混合物中,連續通入1.5 mol/h的R112a以及1.8 mol/h的SO3,反應溫度50℃,一氯二氟乙酰氯的收率可達到96.3%。

常熟振氟專利[12]介紹了將1,1,2-三氯-2,2-二氟乙烷(R122,CClF2-CHCl2)通過紫外光催化氯化生成R112a,再將R112a催化氧化制備氯二氟乙酰氯的方法。例如,在40℃下加入4230 g的R122,通入600 g/h的Cl2,紫外燈光照下反應3 h,R112a的收率為84.7%;將3155 g制得的R112a滴加入3840 g的SO3與35 g的氯磺酸混合物中,2 h滴完,反應溫度控制在50℃,氯二氟乙酰氯的收率為81.3%。
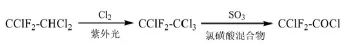
Elf Atochem S.A.專利[13]報道了在氧和化學游離基引發劑存在的條件下使R112a與乙醇反應制備氯二氟乙酸乙酯的方法。在反應器中加入105 g乙醇,150 g含有45 g的R112a的乙醇溶液,加入0.72 g化學游離基引發劑偶氮二異丁腈(AIBN),升溫至65℃,攪拌下通入7 L/h空氣,反應15 h后R112a轉化率為73.4%,氯二氟乙酸乙酯收率為23%。
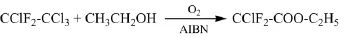
1.1.2 以1,1-二氟四鹵代(含溴/碘)乙烷為原料制備氯二氟乙酰氯
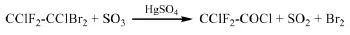
早期德國專利[14]報道了以1,1-二氟-1,2-二氯-2,2-二溴乙烷為原料氧化制備氯二氟乙酰氯的方法。反應溫度為50℃,以65%發煙硫酸為氧化劑,在HgSO4的催化下,1,1-二氟-1,2-二氯-2,2-二溴乙烷氧化生成氯二氟乙酰氯,其中Hg-SO4用量為原料的1wt.%,發煙硫酸用量與原料的重量比為1:1,氯二氟乙酰氯的收率為79%。
1.1.3 以1,1,1-三氯三氟乙烷為原料制備三氟乙酰氯
早期Du Pont專利[15]介紹了在汞鹽催化下以1,1,1-三氯三氟乙烷(R113a,CF3-CCl3)和SO3為原料制備三氟乙酰氯,再用堿水解、硫酸酸化并蒸出三氟乙酸的方法。各物質的份數比為R113a :SO3:(Hg2SO4+HgSO4)=30:40:0.5。Pennsalt Chemicals專利[16]采用相類似的方法,以汞鹽為催化劑,在室溫下進行反應,摩爾比為R113a:SO3= 0.23:0.96,反應5 h,再將生成的三氟乙酰氯水解得到三氟乙酸,其中R113a的轉化率為74.5%。
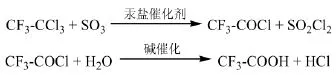
Haloearbon Prod.專利[17]報道了R113a與65%發煙硫酸反應制備三氟乙酰氯的方法,催化劑為HgSO4和Hg2SO4。623 g的R113a與3 g的HgSO4、3 g的Hg2SO4混合,在5 h內緩慢加入753 g的65%發煙硫酸,通過蒸餾可以得到399 g的產物三氟乙酰氯,收率為91%。Kali-Chemie Aktiengesellschaft專利[18]同樣以HgSO4和Hg2SO4的混合物為催化劑,將R113a與液相SO3、SO2Cl2反應制備三氟乙酰氯。在含有4 mol的SO3、0.5 mol的SO2Cl2、0.25 mol的H2SO4和1 g HgSO4、1 g Hg2SO4催化劑的混合物中通入R113a,反應溫度60℃,摩爾比R113a:SO3=1:1~1.3,三氟乙酰氯的收率可達到96%。
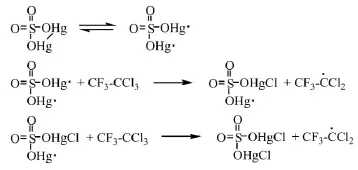
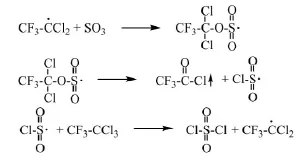
浙江大學劉志政[19]研究了R113a和SO3的反應機理:硫酸亞汞(Hg2SO4)加熱生成硫酸亞汞自由基(O2S(OH·)2),引發R113a生成三氟二氯乙烷自由基(CF3CCl2)。它和三氧化硫結合生成自由基,然后分解生成三氟乙酰氯和硫酰一氯自由基(·SO2Cl),后者引發R113a從而發生自由基連鎖反應。具體反應方程式如下:
(1)自由基鏈引發
(2)自由基鏈增長
(3)自由基鏈終止
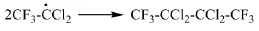
研究認為,較優的工藝操作條件為溫度65℃、摩爾比SO3:R113a=1.1:1、催化劑(Hg2SO4和HgSO4)的質量分數為1%、硫酸的質量分數為2%,在該條件下三氟乙酰氯的收率可達到93%。
浙江大學專利[20]介紹了R113a和SO3在催化劑氟磺酸汞和氟磺酸亞汞的作用下反應制備三氟乙酰氯,同時將副產物SO2Cl2氟化聯產SO2F2的方法。在反應精餾塔中裝填已經交換了汞鹽和亞汞鹽的氟磺酸樹脂填料,通入600 mol/h的R113a、600 mol/h的SO3,控制塔釜溫度120℃、回流比2.5,從塔頂采出三氟乙酰氯的量為594 mol/h。反應生成的SO2Cl2經分離后,與HF在Pd/C催化劑的作用下反應生成SO2F2。
Kali-Chemie Aktiengesellschaft專利[21]采用較為復雜的催化體系對R113a與SO3進行催化制備三氟乙酰氯。催化劑由汞鹽、硼鹵化物及鹵代磺酸等組成,例如,以2 g的HgSO4、17.5 g的HSO3F、2 g的BF3所組成的混合物作為催化劑,與93.7 g的R113a和162.0 g的SO3反應,反應收率沒有報道。
Allied Corporation專利[22]公開了使用鹵素催化劑催化R113a和SO3制備三氟乙酰氯的方法,避免使用有毒有害的含汞催化劑。催化劑包括碘、溴、一氯化碘、一溴化碘、一氯化溴,優選溴催化劑。向帶有攪拌的三口燒瓶中加入150 g的R113a和7.5 g的溴,室溫下30 min內加入145 g的SO3,25℃~60℃反應12 h,R113a的轉化率為80%,三氟乙酰氯的選擇性為98%。
1.2 光氧化法
1.2.1 以R122為原料制備氯二氟乙酰氯
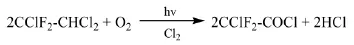
早期Hoechst專利[23]報道了一種在Cl2存在下,在光引發作用下,R122與O2發生光化學氧化連續制備氯二氟乙酰氯的方法。反應溫度120~130℃,R122、O2和Cl2的投料速度分別為140 g/h、28 L/h、0.5 L/h,氯二氟乙酰氯的收率約為82.7%。
Solvay專利[24]報道了R122與O2發生光化學氧化反應,連續制備氯二氟乙酰氯的方法。引發光源為高壓汞蒸氣燈,在溫度100℃、摩爾比R122:O2=1:2.2、停留時間30 min的條件下,R122的轉化率可達到89%,氯二氟乙酰氯的收率為92.9%,副產物主要為COClX、CO2、COF2。在此基礎上,Solvay專利[25]報道了向反應中添加Cl2作為光引發劑,以λ≥280 nm光作為引發光源的方法,在溫度100℃、摩爾比R122:O2:Cl2=1:1.4: 0.24、R122進料量為1.82 mol/h的條件下反應,R122的轉化率可達到94%,氯二氟乙酰氯的選擇性為93%,同時生成少量的R112a。
湖北卓熙專利[26]采用相類似的方法,在高壓汞燈下通入R122與O2、Cl2,發生光化學氧化反應得到氯二氟乙酰氯。選用1000 W的高壓汞燈作為引發光源,在溫度60℃~150℃,摩爾比R122: O2:Cl2=1:0.5:0.35的條件下,氯二氟乙酰氯的收率可達到85%。
1.2.2 以1,1-二氯-2,2-二氟乙烯為原料制備氯二氟乙酰氯
Hoechst專利[29]報道了在紫外燈的照射下,1,1-二氯-2,2-二氟乙烯(R1112a,CF2=CCl2)光氧化制備氯二氟乙酰氯的方法。反應溫度120℃~130℃,停留時間9 min,氯二氟乙酰氯的收率為80%。在此基礎上,Solvay專利[30]使用高壓汞燈作為引發光源,通過入≥280 nm光引發進行R1112a的光氧化反應。在85℃~91℃的溫度下,0.6 mol的R1112a與1.0mol的O2、0.11mol的Cl2反應20min,R1112a的轉化率達到99.58%,氯二氟乙酰氯的選擇性為90.8%。
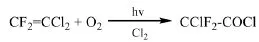
1.2.3 以1,1-二氯-2,2-二氟乙烷為原料制備二氟乙酰氯
Asahi Glass公司專利JP08053388A[29]介紹了一種以1,1-二氯-2,2-二氟乙烷(R132a,CHF2-CHCl2)為原料光氧化合成二氟乙酰鹵及其二氟乙酸酯的制備方法。在80℃條件下,將摩爾比R132a:O2:Cl2=1:3:0.5的混合物用400 W高壓汞燈照射5 min,得到二氟乙酰氯,R132a的轉化率為99%,二氟乙酰氯選擇性為99%。二氟乙酰氯進一步水解可以得到二氟乙酸。
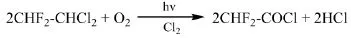
1.2.4 以2,2-二氯-1,1,1-三氟乙烷為原料制備三氟乙酰氯
Haszeldine等[30]提出了氣相光氧化1,1,1-三氯三氟乙烷(R113a,CCl3-CF3)、2,2-二氯-1,1,1-三氟乙烷(R123,CF3-CHCl2)、2-氯-1,1,1-三氟乙烷(R133a,CF3-CH2Cl)分別制備三氟乙酰氟、三氟乙酰氯和三氟乙酸的方法,認為通過添加光引發劑Cl2可以大幅提升目標產物的收率。
Pennsalt Chemicals專利[31]描述了通過加熱,紫外光、γ光線、X射線或高能電子輻射等方式引發自由基,將含鹵乙烷CF2MCXYZ(其中,M=F或Cl,X,Y=H,Cl或Br)氧化生成相應含鹵乙酰氯或含鹵乙酸的方法。在光引發劑Cl2存在的情況下,采用加熱的方式,反應溫度優選200℃~250℃;采用紫外光或其它輻射的方式,則反應溫度可以在-30℃以下。專利實施例中,在紫外光輻射下,通入摩爾比為R123:O2:Cl2=1:4:2的物料,在2個大氣壓的條件下反應,R123可達到完全轉化,生成的產物中絕大多數為三氟乙酰氯。
Halocarbon Products專利[32]描述了通過紫外燈輻射氣相氧化R123制備三氟乙酰氯的方法。將含有2%雜質CFCl2-CF2Cl的R123、O2分別以45.36 kg/h、5.44 kg/h的速率通入反應器中,以2000 W紫外燈作為光源,在溫度105℃、壓力120 psig(即0.83 MPa)的條件下反應,R123的轉化率為95%,三氟乙酰氯的收率可達到99%。
Du Pont專利[33]介紹了在Cl2存在下通過液相氧化R123或者R133a制備三氟乙酰氯的方法。向裝滿R123的1.6 L玻璃反應器中持續1.5 h通入80 mL/min的O2,接著添加Cl2直至溶液中氯的濃度達到0.0033 mol/L,打開450 W、波長>280 nm的中壓汞燈,通入O2、Cl2進行反應,期間O2的流速為80 mL/min,保持溶液中氯的濃度≤0.0035 mol/L,可以獲得純度為99.0%的三氟乙酰氯。研究認為,當輻射光波長≤280 nm,容易導致副產物HF的生成;而在Cl2存在的情況下,使用波長>280 nm的高壓或中壓汞燈光源,有助于提高反應的選擇性,同時可以避免玻璃反應器的腐蝕問題。
Solvay專利[34]報道了R123與氧氣發生光氧化反應連續制備三氟乙酰氯的方法。選用700 W的高壓汞蒸汽燈作為引發光源,在100℃、摩爾比R123:O2=1:2.28、停留時間30 min的條件下,R123的轉化率達到95%,三氟乙酰氯的收率為85%,副產物主要為COCl2、CO2、COF2。在此基礎上,Solvay專利[35]報道了向反應中添加Cl2作為光引發劑的方法,選用500 W的高壓汞蒸汽燈作為引發光源,通過濾光片濾去λ<280nm的光,在溫度100℃、摩爾比R123:O2:Cl2=1:1.23:0.2、R123的進料量為1.92 mol/h的條件下,R123的轉化率接近100%,三氟乙酰氯的選擇性為100%。
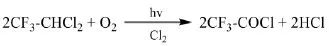
杭州原正化工專利[36]介紹了將三氟乙烷氯化混合物與O2、Cl2在汞燈輻射下進行光氧化反應制備三氟乙酰氯的方法。向反應釜中加入5 L的三氟乙烷氯化混合物(9%的R133a、90%的R123、1%的R113a),攪拌下連續通入500 mL/min的O2、250 mL/min的Cl2,反應溫度5℃,壓力0.09 MPa,汞燈輻射,共生成10.3 mol的三氟乙酰氯。該公司專利CN101735033A[37]、CN101735034A[38]介紹了在Cl2存在的情況下,R123與O2進行液相光氧化反應制備三氟乙酰氯的方法。為防止生成的三氟乙酰氯被汞燈輻射分解,分別采用了在物料泡點溫度或泡點溫度以上進行反應,在反應器氣液相界面以下加裝遮光構件的方法。
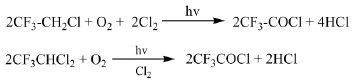
1.2.5 以R113a為原料制備三氟乙酰氯
Pennsalt Chemicals專利[39]介紹了以紫外線為引發光源,R113a與O2在Cl2存在的條件下反應制備三氟乙酰氯的方法。當反應中存在水時,生成的三氟乙酰氯進一步反應生成三氟乙酸。例如,將摩爾比為R113a:O2:Cl2=5:2:3的物料在2個大氣壓的條件下反應72 h,當存在水時,三氟乙酸的收率為50%。
Daikin專利[40]介紹了以紫外線為引發光源,R113a與SO3或發煙硫酸在Br2存在的條件下反應制備三氟乙酰氯的方法。60℃、常壓下反應,紫外燈功率為20 W,三氟乙酰氯的收率可達到90.1%。當沒有紫外燈照射時,反應產物幾乎沒有。
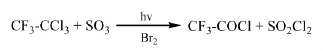
1.3 催化氧化法
1.3.1 以1,2-二氯-1,1,2-三氟乙烷為原料制備氯二氟乙酰氯
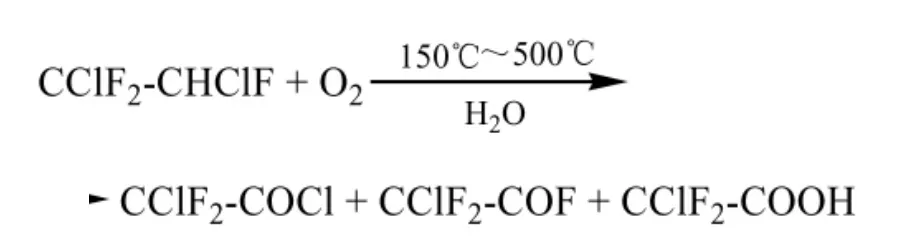
Asahi Glass專利[41]報道了1,2-二氯-1,1,2-三氟乙烷在少量水的存在下,與O2發生熱氧化反應,反應溫度優選150℃~500℃,得到CClF2-COCl、CClF2-COF及CClF2-COOH混合物,該混合物與水反應,得到CClF2-COOH。
1.3.2 以R123為原料制備三氟乙酰氯
Asahi Glass專利[42]介紹了在水存在的條件下R123氣相氧化制備三氟乙酰氯和三氟乙酸的方法。向反應器中通入R123、O2、H2O,R123的流量為103 mol/h,物料摩爾比R123:O2:H2O=1:1: 0.1,在溫度300℃、壓力30 kg/cm2、停留時間8.4 min的條件下反應,通過攪拌槳攪拌避免反應器內局部過熱,R123的轉化率為95%,三氟乙酰氯的選擇性為68%,三氟乙酸的選擇性為26%。在該反應中,水起到了催化劑的作用,但不可避免地將R123氧化得到的三氟乙酰氯水解生成三氟乙酸,而水與三氟乙酰氯、三氟乙酸分離較為困難。此外,水的含量過高會導致其與氟化氫形成氫氟酸而腐蝕設備。
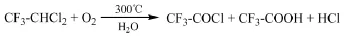
Du Pont專利[43]介紹了通過活性炭催化氧化R123制備三氟乙酰氯的方法。將R123、O2分別以0.10 mL/min、25 mL/min的速率通入裝填有活性炭催化劑的反應管,活性炭的比表面積為600~1500 m2/g,反應溫度275℃、停留時間16 s,R123的轉化率可達到100%,但三氟乙酰氯的選擇性僅為39%,其它副產物包括R113a、R13、R23,這可能是由于較高的反應溫度導致部分原料中CC鍵的斷裂。
寧丹等[44]以活性炭為催化劑,在連續流動固定床反應器上將R123催化氧化制備三氟乙酰氯。研究認為,反應的轉化率和選擇性隨著活性炭比表面積和孔容增大而增加,活性炭表面的酸性也有助于其催化性能的提高。例如,在比表面積921 m2/g的活性炭催化下,反應溫度270℃~280℃,物料摩爾比R123:O2=0.65:1,HCFC-123空速0.55 h-1,三氟乙酰氯最高收率為71.1%。

1.4 熱分解法
1.4.1 以1,3-二氯-1,1,3,3-四氟丙酮為原料制備氯二氟乙酰氯
Allied Chemical&Dye專利[45]報道了1,3-二氯-1,1,3,3-四氟丙酮熱分解制備氯二氟乙酰氯及四氟乙烯的方法,選擇的熱解溫度范圍為500℃~750℃。例如,將原料汽化后通入反應器,流速2.00 mol/h,在675℃、停留時間0.6 s的條件下進行熱解,反應轉化率為37%,氯二氟乙酰氯的收率為72%。

1.4.2 以1-烷氧基-1,1,2,2-四氟乙烷為原料制備二氟乙酰氯
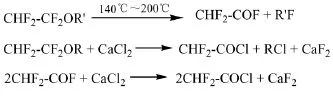
中央硝子專利[46]介紹了由1-烷氧基-1,1,2,2-四氟乙烷(ATFE)熱分解得到的二氟乙酰氟高效率地轉變為二氟乙酰氯的方法。首先將ATFE在溫度140℃~200℃、反應時間10~300 s的條件下催化熱分解生成二氟乙酰氟及氟代烷;將蒸餾得到的二氟乙酰氯通入填充有粒狀無水CaCl2的反應管中,反應溫度160℃時得到的氯化產物中,二氟乙酰氯的含量為93.7%。
1.5 其它方法
1.5.1 三氟乙醛光氯化制備三氟乙酰氯
Hoechst AG專利[47]介紹了將Cl2通入液相三氟乙醛中,通過光氯化制備三氟乙酰氯的方法。將124份的Cl2通入裝有162份三氟乙醛的反應器中,在紫外燈照射下,-30℃進行反應,三氟乙酰氯的收率為87%。此反應過程不易控制,通入Cl2較為危險。
1.5.2 R123超臨界氧化制備三氟乙酰氯
Du Pont專利[48]介紹了在超臨界狀態下氧化R123制備三氟乙酰氯的方法。將41.3 g的R123通入充填有O2,壓力為120 psig(即0.83 MPa)的反應器中,在220℃、700 psig(即4.8 MPa)的條件下反應15 min,R123的轉化率為90%,三氟乙酰氯的選擇性為91%,反應中有副產物HF、三氟乙酸、R113a產生。由于超臨界狀態下的高溫高壓對反應器要求苛刻,工業上較難實現,不適合大規模生產。
2 結論與展望
作為一類重要的脂肪族含氟中間體,含氟乙酰氯類化合物的經濟價值已日益凸顯。其合成技術的選擇與發展方向如下:
SO3氧化法的主要優點是可以在較低的反應溫度和常壓下進行,反應條件比較溫和,對設備的要求不是十分苛刻,主要缺點是需采用SO3等危險化學品和有毒的汞鹽作催化劑,反應過程中產生的硫酰氯的分離和利用問題也有待解決。
光氧化法,即在紫外光照射下,氯氟烷烴/烯烴和O2在一定溫度和壓力下反應制得相應的含氟酰氯。該方法反應條件較為溫和,在反應中加入少量的鹵素化合物,如Cl2、Br2等,可提高反應速度。值得注意的是,光氧化反應器的設計是該方法實現工業化的關鍵,需避免光照引起的產物分解問題。
催化氧化法需要在高溫條件下進行,研究主要集中在水和活性炭兩類催化劑。由于會產生氫氟酸的腐蝕問題,在工業生產中采用水催化的氧化反應是不合適的;而開發的活性炭催化劑,則會使反應的副產物較多,仍需進一步研究改進。
熱分解法的反應條件較為苛刻,并且需要特定的原料,目前研究相對較少。
[1]徐衛國,陳先進,徐宇威,等.一氯二氟醋酸的合成與應用[J].浙江化工,2004,35(6):20-21.
[2]趙衛娟,徐衛國,劉毓林,等.一氯二氟乙酰氯及其衍生物的制備及應用[J].浙江化工,2013,44(7):1-3.
[3]Ooharu,Kazuya,Kumai,Seisaku.Preparation of difluoroacetyl halides and difluoroacetic acid:JP,08053388[P]. 1996-12-27.
[4]Olivier B,Roland J.Production of difluoroethanol:US, 7902409B2[P].2011-03-08.
[5]陳偉,徐衛國,李華,等.二氟醋酸的合成方法研究[J].有機氟工業,2014,(1):43-46.
[6]Gryszkiewicz-Trochimowski M E,Sporzyn?ski A,Wnuk J.Recherches sur les Composés Organiques Fluorés dans la Série Aliphatique:II.Sur les dérivés des acides mono-, di-et tri-fluoro-acétiques[J].Recueil des Travaux Chimiques des Pays Bas,1947,66(7):419-426.
[7]Tinker J M.Trifluoroacetyl halide and a process of making it:US,2257868[P].1941-10-07.
[8]Henne A L.Alm R M,Smook M.Trifluoroethanol[J].J. Am.Chem.Soc.,1948,70(5):1968.
[9]Saunders J H,Slocombe R J,Hardy E E.The preparation of α-trifluoro-p-phenylacetophenone[J].J.Am.Chem. Soc.,1949,71(2):752.
[10]Dittman A L,Zager R I.Preparation of perfluoro-and perchlorofuoro-acetyl chloride:US,3160659[P].1964-12-08.
[11]Paucksch H,Massonne J,Bohm H,et al.Process of preparing fluorine containing perhalogencarboxylic acid fluorides or chlorides:US,3725475[P].1973-04-03.
[12]沈達.一種三氟乙酸的制備方法:CN,103524325[P]. 2014-01-22.
[13]Drivon G.,Gillet J P,Ruppin C,et al.Process for the preparation of alkyl halodifluoroaceates:US,5619023[P]. 1997-04-08.
[14]Dieter G.Verfahren zur herstellung von perhalogencarbonsaurefluoriden order-chloriden,deren halogengehalt zu mehr als 50 molprozent aus fluor besteht:DE,1020970B [P].1957-12-19.
[15]Benning A F,Park J D.Preparation of trifluoro acetic acid: US,2396076[P].1946-05-05.
[16]Lawlor F E,Braid M.Oxidation process for preparing car-boxylic acid anhydrides:US,3102139[P].1963-08-27.
[17]Dittman A L,Zager R I.Preparation of perfluoro-and perchlorofuoro-acetyl chloride:US,3160659[P].1964-12-08.
[18]Paucksch H,Massonne J,Bohm H,et al.Process of preparing fluorine containing perhalogencarboxylic acid fluorides or chlorides:US,3725475[P].1973-04-03.
[19]劉志政.CF3CCl3衍生物的合成及反應機理研究[D].杭州:浙江大學,2012.
[20]尹紅,袁慎峰,陳志榮,等.一種連續合成三氟乙酰氯和硫酰氟的方法:CN,102351681A[P].2012-02-15.
[21]Rudolph W,Fernschild G,Hirsch W.Process for preparing trifluoracetylchlorid:EP,0029591A1[P].1980-11-21.
[22]Anello L G,Eibeck R E,Robinson M A.Process for the preparation of perhaloalkanoyl chloride:US,4340548[P]. 1982-07-20.
[23]Otto S,Heinrich K,Heinrich M.Verfahren zur herstellungvon fluor und chlor enthahenden essigssaure chloride: DE,1069137[P].1958-01-10.
[24]Braun M,Rudolph W,Einchholz K.Process for preparing polyfluorocarboxylic acid chlorides and perfluotocarboxylic acid chlorides:US,5545298[P].1996-08-13.
[25]Braun M,Rudolph W,Einchholz K.Process for preparing polyfluorochlorocarbonyl chlorides and perfluorocarbonyl chlorides with addition of chlorine:US,5569782[P].1996-10-29.
[26]謝學歸,張雨寒.一種一氯二氟乙酰氯的制備方法:CN, 103351292A[P].2013-10-16.
[27]Otto S,Heinrich K,Heinrich M.Verfahren zur herstellung von fluor und chlor enthahenden essigssaure chloride:DE, 1069137[P].1958-1-10.
[28]Braun M,Rudolph W,Eichholz K.Difluorochloracetyl, dichloracetyl and trichloracetyl chloride preparation:US, 5919341[P].1999-07-06.
[29]Oharu K,Kumai S.Production of difluoroacetic acid halide and difluoroacetic acid:JP,08053388A[P].1996-02-27.
[30]Haszeldine R N,Nyman F.Oxidation of polyhalogenocompounds.PartⅡ.Photolysis and photochemical oxidation of some chlorofluoroethanes[J].J.Chem.Soc.,1959, 387-396.
[31]Braid M,Lawlor F.Synthesis of fluorine compounds:US, 3151051[P].1964-09-29.
[32]Dittman A L.Production of 2,2,2-trifluoroacetyl chloride: US,3883407[P].1975-05-13.
[33]Huang H N.Process for omega-halo-perfluoro acid chlorides:US,5259938[P].1993-11-09.
[34]Braun M,Rudolph W,Einchholz K.Process for preparing polyfluorocarboxylic acid chlorides and perfluotocarboxylic acid chlorides:US,5545298[P].1996-08-13.
[35]Braun M,Rudolph W,Einchholz K.Process for preparing polyfluorochlorocarbonyl chlorides and perfluorocarbonyl chlorides with addition of chlorine:US,5569782[P].1996-10-29.
[36]趙建明,韓箴賢,崔覺劍.一種由三氟乙烷氯化混合物制備三氟乙酰氯的方法:CN,101747176A[P].2009-12-24.
[37趙建明,韓箴賢,崔覺劍.一種由2,2-二氯-1,1,1-三氟乙烷制備三氟乙酰氯的方法:CN,101735033A[P].2009-12-18.
[38]趙建明,韓箴賢,崔覺劍.一種三氟乙酰氯的制備方法: CN,101735034A[P].2009-12-18.
[39]Braid M,Lawlor F.Synthesis of fluorine compounds:US, 3151051[P].1964-09-29.
[40]Yoshida T,Kanetani T,Misaki S.Preparation of perhalogenoalkanoic acid chloride:JP,60237040A[P].1984-05-09.
[41]Kumai S,Seki T.Production of chlorodifluoroacetic acid: JP,6239792A[P].1994-08-30.
[42]Gotoh I,Yoneda H,Kumai S,et al.Process for producing trifluoroaceticacidandtrifluoroacetylchloride:US, 5041647[P].1991-08-20.
[43]Jacobson S E.Process for producing trifluoroacetyl chloride:US,5241113[P].1993-08-31.
[44]寧丹,黃晴,吳銀登,等.活性炭在催化氧化三氟二氯乙烷制備三氟乙酰氯中的應用[J].工業催化,2007,15(2): 62-64.
[45]Miller C B,Cyril W.Manufacture of fluorochloroacetyl halide:US,2741634[P].1956-04-10.
[46]岡本正宗,井村英明,高田直門.二氟乙酰氯的制造方法: CN,102822134A[P].2012-12-12.
[47]Scherer O,Hahn H.Process for preparing trifluoroacetyl chloride:US,3320142[P].1967-05-16.
[48]Jacobson S E,Ely W B.Process for preparing perhaloacyl chlorides:US,5296640[P].1994-03-22.
Research Progress in the Synthesis of Fluorinated Acyl Chlorides
ZHANG Di1,WANG Shu-cheng1,WANG Jun-xiang2,LIN Sheng-da1,YU Wan-jin1,LIU Wu-can1
(1.Zhejiang Chemical Industry Research Institute,The National ODS Substitutes Engineering&Technology Research Center,Hangzhou,Zhejiang 310023,China;2.Xiasha Production Base of Sinochem Lantian Co.,Ltd.,Hangzhou,Zhejiang 310018,China)
Several methods for the synthesis of fluorinated acyl chlorides,include SO3-oxidation,photooxidation,catalytic oxidation,thermal decomposition and so on,were introduced and compared.Furthermore, the synthetic trend of fluorinated acyl chlorides was considered.
chlorodifluoroacetyl chloride;2,2-difluoroacetyl chloride;trifluoroacetyl chloride;photo-oxidation;catalytic oxidation
1006-4184(2015)2-0005-07
2014-05-07
張迪(1986-),女,碩士,工程師,2011年7月畢業于浙江工業大學,目前主要從事氟化催化劑及ODS替代品方面的研究。E-mail:zhangdi1@sinochem.com。
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學生數理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
