CP/MAS 13C NMR光譜法分析煙草纖維素的結構
2015-11-24 07:13:37馮廣林譚蘭蘭朱曉蘭高蕓戴亞
中國煙草學報 2015年5期
關鍵詞:煙草
馮廣林,譚蘭蘭,朱曉蘭,高蕓,戴亞
1 川渝中煙工業有限責任公司技術研發中心, 成都 610017;2 中國科學技術大學煙草與健康研究中心,合肥 230052
煙草與煙氣化學
CP/MAS13C NMR光譜法分析煙草纖維素的結構
馮廣林1,譚蘭蘭1,朱曉蘭2,高蕓2,戴亞1
1 川渝中煙工業有限責任公司技術研發中心, 成都 610017;2 中國科學技術大學煙草與健康研究中心,合肥 230052
采用交叉極化/魔角旋轉 (CP/MAS)13C NMR光譜法結合光譜擬合技術對酸性洗滌劑洗脫的煙草纖維素樣品進行了晶體結構分析,光譜峰的擬合結果顯示煙草纖維素晶型以Iβ為主要晶體形式,煙草纖維素的結晶度在50%左右。煙草纖維素基原纖尺寸在3.0-5.0 nm之間,基原纖聚集束的尺寸在6-13 nm。此項研究可以為纖維素改性、煙草加工以及低次煙葉和煙梗的處理等方面提供重要的理論依據。
煙草;纖維素; CP/MAS;NMR光譜;結晶結構
纖維素作為構成煙葉細胞組織和骨架的基本物質,在煙葉中含量一般在11%左右,并隨著煙葉等級的下降而增加。纖維素可以提高煙葉燃燒性,使煙葉持火力增強,但纖維素含量過高時,煙葉組織粗糙而容易破碎[1]。在低次煙葉中,還原糖和可溶性總糖含量很低,纖維素、半纖維素含量較高,致使低次煙葉具有強烈的刺激味、青雜氣重、吸味辛辣、澀口,在煙葉燃吸時會引起刺激性的嗆咳。因此,煙草配方中高纖維素含量,會對燃吸品質產生副作用,賦予煙氣一種尖刺的刺激性和一種“燒紙”的氣味,使煙葉的香氣不能顯露,影響煙葉的感官質量[2]。在煙草的加工過程中,纖維素大分子晶型結構及原纖尺寸的變化等因素對于煙草的潤脹吸濕性能和柔韌性等品質都有重要的影響[3]。因此,纖維素含量及其微觀結構對于煙葉的品質和利用有很重要的意義。
對于纖維素的分析測定工作大致可以分為三個階段:粗纖維法、纖維洗滌劑法和酶法[4]。目前的行業標準方法[5]是采用中性洗滌劑、酸性洗滌劑以及72%硫酸分別依次除去煙草中的非細胞壁物質,并經過濾、洗滌、干燥、灰化、稱重、計算等步驟分別得到中性洗滌纖維、酸性洗滌纖維和酸洗木質素的含量,進而換算得到纖維素的含量。該方法雖然具有選擇性好、靈敏度高的特點,但操作比較繁瑣,操作周期長、工作量大,更為重要的是對煙草中此類構架類物質的結構和組成沒有涉及,對纖維素的晶型組成及比例的研究也基本空白[6]。
交叉極化結合魔角旋轉13C核磁共振法(CP/MAS13C NMR)技術由于能夠直接采用固體粉末定量地估計其化學組成和化學結構,對其組分不必進行分離和溶解等處理就可以直接測定而獲得可靠的結構信息,已經成為研究高分子化合物化學結構和物理性質最重要的工具之一[7-9]。本論文采用CP/MAS13C NMR法結合光譜擬合技術研究煙草纖維素晶體的晶型分布及比例、纖維素的結晶度、原纖聚集態尺寸等結構信息,對于進一步研究其對煙草加工和潤脹性能的影響,改善煙葉的吸食質量,提高低次煙葉和煙梗的使用價值具有重要應用價值和現實意義。
1 材料與方法
1.1 試劑與材料
正辛醇(C8H18O,消泡劑)、十六烷基三甲胺溴(C19H42BrN)、98%濃硫酸(質量分數)、石英砂、乙醇、丙酮、苯(AR,上海化學試劑四廠)。纖維素標樣(純度>98%,上海百靈威化學品公司)。水為二次雙蒸水。
酸性洗滌劑(2%十六烷基三甲胺溴):用50 mL量筒量取約27.2 mL 98%濃硫酸,緩慢加入已裝有500 mL蒸餾水的燒杯中,冷卻后轉移至1000 mL容量瓶,使用水定容至刻度,配置成1.0 mol/L硫酸溶液;稱取約20 g十六烷基三甲胺溴于1000 mL燒杯中,加入配置好的1.0 mol/L硫酸溶液,攪拌溶解,轉移至1000 mL容量瓶,定容至刻度。
煙葉樣品取自川渝中煙工業公司,包括云南烤煙、湖南香料煙、湖北白肋煙和廣州曬黃煙4個不同類型及貴州畢節上、中、下3個部位不同等級(B2F、C3F和X2F)烤煙煙葉,煙樣于40 ℃烘箱中干燥4 h后,粉碎,過150 μm(100目)篩,備用。
小型設備有HHS11-2型恒溫水浴鍋;D2010W型電動攪拌器;SL234型電子天平(感量:0.0001 g,美國Denver公司);PHS3C型精密pH計;FZ102型植物粉碎機;TNX1600-30馬弗爐、BridgeLux纖維分析儀(配置石英過濾坩堝)。
1.2 煙草纖維素的提取和分離[5]
取兩份煙樣,按照行業標準(YC/T 347—2010)進行相同的前處理,即采用上述1.3的酸性洗滌劑溶劑和72%硫酸溶液洗脫煙草有機溶劑溶解物如糖、淀粉、蛋白質、果膠及半纖維素等得到纖維素,一份進一步灰化計算出含量(纖維素含量為9.84%,木質素2.87%),另一份作為煙草纖維素參比樣品,進行CP/MAS13C NMR分析。此樣品的光譜圖扣除根據灰化測定的纖維素含量的制備的纖維素標準樣品光譜吸收,得到煙草樣品的非纖維素背景光譜吸收。待測煙樣同樣采用行標方法洗脫后,其CP/MAS13C NMR光譜扣除非纖維素背景光譜吸收即可得到待測樣品的纖維素光譜圖。
1.3 儀器及測試方法
1.3.1 煙草纖維素的CP/MAS 13C NMR光譜分析
CP/MAS13C NMR光譜利用AVANCE AV 400 spectrometer超導傅立葉數字化核磁共振譜儀(瑞士Bruker)記錄,測試條件:場強9.40 T,4 mm和7 mm魔角探頭,轉速15 kHz,脈沖寬度90°,交叉極化時間4 μs,接觸時間2 ms,采樣時間34 ms,采樣間隔時間2.0 s,掃描次數:1024,譜寬300 ppm,一級定標用四甲基硅烷(CH3)4Si。13C CP/MAS NMR光譜化學位移、光譜峰面積及在δ 60-110 ppm的分解峰形擬合的計算是采用MestReNova 6.1.1核磁軟件進行分析,峰形分解的結果用來計算纖維素的晶型分布及比例、纖維素的結晶度、原纖聚集態尺寸等結構信息。
1.3.2 煙草纖維素的XRD光譜分析
XRD分析在日本瑪珂公司TTR-Ⅲ型大功率X射線粉末衍射儀上進行測定,纖維素樣品安放在玻璃樣品架上,在穩定條件下分析。測試條件:Ni濾波,Cu靶Kα射線,管壓 40 kV,管流200 mA,掃描速度 2(o)/min,掃描范圍從 3 o-50 o。波長:1.541841 A。
1.3.3 煙草纖維素的紅外光譜分析
KBr紅外光譜分析是將煙草纖維素樣品在美國Nicolet MAGNA-IR8700型傅里葉變換紅外光譜儀上進行測定,光譜儀分辨率為0.1 cm-1,掃描范圍:4000-400 cm-1,掃描次數為50次,掃描速度:0.1 cm/s。
2 結果與討論
2.1 煙草纖維素分子CP / MAS 13C NMR光譜分析
在對轉速、魔角探頭、交叉極化時間和弛豫時間等參數進行優化后,最終采用1.3.1所示的實驗參數條件,對纖維素標準樣品及煙草纖維素樣品進行核磁共振波譜分析。圖1為纖維素樣品CP/MAS13C NMR光譜,樣品分別為纖維素標樣、酸性洗滌劑洗脫和熱蒸餾水洗脫的煙草纖維素樣品,光譜吸收所對應的化學位移歸屬如表1。煙草纖維素的13C NMR吸收信號主要在化學位移δ60-110 ppm處[10], 95-110 ppm處吸收峰對應的是纖維素的C1區,表征的是晶態結構,通過擬合可以得到纖維素的晶型及其比例,化學位移80-90 ppm C4區對應的是纖維素的結晶區和無定型區兩個部分,可以此研究纖維素結晶區內和結晶基原纖表面及無定型區的纖維素分子排列情況,因此擬合結果可以計算結晶度和原纖尺寸。化學位移63 ppm 和68-76 ppm吸收峰分別對應的是C-6和吡喃環上其他C(C-2,3,5)。另外,還有一些弱的雜質吸收峰,如53 ppm處的甲氧基吸收和30 ppm處的脂肪碳吸收。如圖1所示,兩種煙草纖維素樣品中,蒸餾水洗脫樣品的光譜因為含有果膠、半纖維素等雜質,譜圖干擾多,所得纖維素樣品純度不高。酸性洗滌劑洗脫樣品光譜圖上C-1吸收峰峰的位置穩定、峰形對稱性較好(有少量木質素雜質),在背景扣除(消除干擾)后其峰面積與纖維素的存在量呈良好的線性關系,可實現煙草樣品中纖維素固體的直接光譜分析[11]。

圖1 煙草提取纖維素樣品的13C CP/MAS NMR光譜圖Fig.1 13C CP/MAS NMR spectra of cellulose samples extracted from tobacco

表1 纖維素分子CP/MAS 13C NMR光譜的化學位移歸屬Tab.1 CP/MAS 13C NMR chemical shifts (in ppm) for cellulose
2.2 煙草纖維素樣品干擾的扣除
如圖1示,按照行業標準方法(酸性洗滌劑法)得到煙草纖維素樣品由于仍然含有木質素等雜質,其13C NMR波譜與纖維素標樣相比在化學位移105ppm纖維素C-1吸收峰處有少量干擾,因此需扣除木質素等干擾吸收峰。如圖2所示,選擇煙草纖維素參比樣品(圖2b)扣除相同含量(按照行業標準方法洗滌灰化后測得纖維素含量)的纖維素標準樣品(圖2a)的光譜吸收后作為非纖維素背景吸收(如圖2c所示),選擇參比樣品的原則是要使非纖維素背景吸收中化學位移105ppm和88ppm處(分別對應C-1 和C-4吸收峰)強度最低但不能出現負峰[11]。最后再將煙草纖維素待測樣13C NMR圖(圖2d)扣除非纖維素背景吸收,得到樣品中僅纖維素吸收的光譜圖(圖2e),再進行后續的結構分析(包括晶型比例、結晶度及原纖尺寸等)。

圖2 煙草酸性洗滌劑提取纖維素樣品及背景吸收的13C CP/MAS NMR光譜圖Fig.2 CP/MAS 13C NMR spectra of tobacco cellulose samples and backgroud
2.3 煙草纖維素的晶型與含量
天然纖維素(包括煙草纖維素)是以纖維素I形式存在的,是纖維素Iα和纖維素Iβ兩種晶體的混合物[12]。纖維素Iα和Iβ分別指的是1個鏈的三斜單元晶胞和2個鏈的單斜單元晶胞,在一定條件下它們可以相互轉化[13]。次晶表示的是有序性不及但運動性大于纖維素Iα和Iβ的結晶結構[3,14],其含量變化可以用來反映晶體結構的變化。
根據Wickholm等的研究結果[12],在CP/MAS13C NMR光譜圖上,對煙草纖維素C1信號峰擬合,采用4個Lorentzian線形表示不同晶型,分別為結晶區纖維素Iα(δ105.1ppm)、纖維素Iβ(δ105.7和δ104.0ppm的雙峰)以及次晶纖維素(δ104.6ppm),根據擬合分峰的面積計算出纖維素Iα和Iβ以及次晶的的相對含量(如圖3和表2示)。

圖3 煙草纖維素樣品的C1區光譜擬合的結果Fig.3 Fitting of C-1 spectral region of 13C CP/MAS NMR spectra of cellulose in tobacco

表2 由13C CP/MAS NMR C1區光譜計算出煙草纖維素樣品(貴州中部烤煙)的晶型含量Tab.2 Contents of crystalline allomorphs obtained from 13C CP/MAS NMR spectra of cellulose in tobacco ( fl ue-cured tobacco from middle area of Guizhou province)
表2顯示了煙草纖維素中各晶型的相對含量,計算得出其Iα和Iβ兩種晶型相對含量的比值為0.425。晶型的組成與植物的種類關系密切,不同來源的纖維素Iα和Iβ晶型含量比值不同。桉木漿纖維、苧麻、棉纖維和竹漿中Iα和Iβ兩種晶型相對含量的比值分別為 0.51,0.47,0.26 和 0.25[15]。
實驗進一步分析了國產烤煙煙葉和煙梗、貴州烤煙不同部位及國產不同類型的煙葉纖維素樣品的晶型含量和分布,結果如表3中所示,所有的樣品均顯示煙草纖維素以Iβ晶型為主,煙梗的纖維素晶型含量和分布與上部煙葉類似。
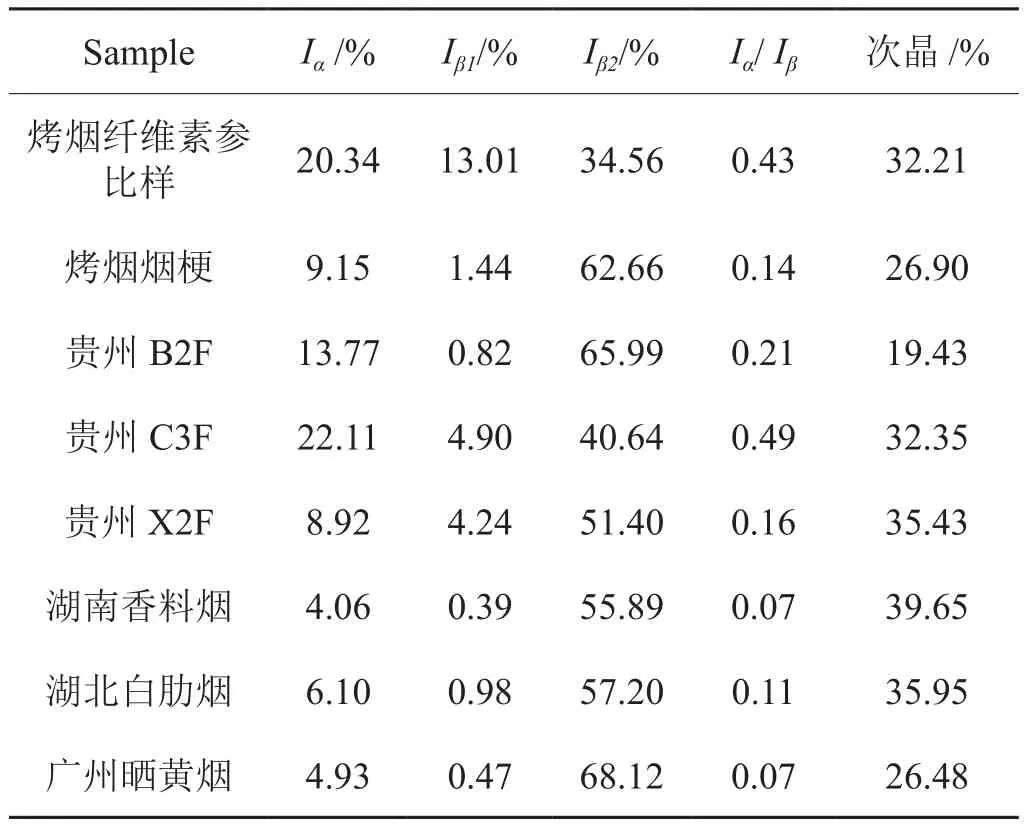
表3 由13C CP/MAS NMR光譜計算的不同煙草纖維素樣品的晶型含量結果Tab.3 Contents of crystalline allomorphs obtained from 13C CP/MAS NMR spectra of cellulose in tobacco samples
2.4 煙草纖維素的結晶度
纖維的結構既不是完全晶態,也不是完全無定型態的,是部分結晶和部分取向,聚集態結構中的結晶部分及無定型部分都是決定纖維素性質的重要方面[16]。纖維素的化學位移80-90 ppm處的C4譜線裂分為兩部分(如圖4):窄的低場和寬的高場,分別對應著纖維素的結晶區和非結晶區,在結晶區的化學位移為δ86-92ppm,在非結晶區為δ80-86ppm,可據此處信號峰的面積按表4中公式來計算纖維素的結晶度[15]。
表4中列出了采用三種不同方法來計算煙草纖維素樣品的結晶度,從結果來看,煙草纖維素樣品的結晶度在50%左右,用13C NMR方法得到的結晶度要比X射線衍射法(XRD法)的計算結果略低些,這是由于相比XRD,13C NMR對小范圍的有序更為敏感,在13C NMR中,結晶區信號峰的強度取決于每個相內部的C原子數目,即只有晶區內的部分才能被看作是結晶區,因而結晶度的大小取決于晶粒尺寸以及結晶的完整性,而提取過程會使纖維結晶體的完整性受到破壞,晶粒的表面積增加。因此,對于結晶區有缺陷的纖維素來說,13C NMR可能更能準確地分析纖維素大分子排列的有序情況[15]。至于紅外光譜法(IR法)測定的結果更高,可能存在少量的木質素干擾,使得結果不夠準確。
不同部位的貴州烤煙樣品結晶度結果顯示下部煙的纖維素結晶度最高,中部煙次之,上部最低。從不同類型煙葉結晶度結果來看,曬黃煙的結晶度最高(54.6%),湖北白肋煙的結晶度最低(46.3%),烤煙與香料煙差距不大。

圖4 煙草纖維素樣品(貴州B2F)的光譜分布Fig.4 CP/MAS 13C NMR spectra of cellulose in tobacco (Guizhou B2F).

表4 由CP/MAS 13C NMR光譜計算出不同煙草纖維素樣品的結晶度Tab.4 Degrees of crystalline obtained from CP/MAS 13C NMR spectra of cellulose in tobacco samples
2.5 煙草纖維素原纖結構的研究
植物纖維細胞壁中纖維素分子鏈平行排列形成基原纖,再由若干根基原纖組成微原纖,微原纖平行的組合在一起形成原纖。微原纖是纖維的主要結構單元,晶區位于微原纖內,稱為“微晶”或“膠束”。基原纖即絲狀多晶體,是結晶纖維素中最小的結構單元。因此通過計算纖維素晶粒尺寸可得到基原纖的橫向尺寸。研究發現,纖維素不同組織水平和結構水平上的變化(纖維、原纖、微原纖、基原纖及纖維素鏈等發生結構的變化)會影響纖維素在化學改性中的活性、纖維素被酶水解的易感性和纖維的強度性能等[18]。
以圖4為基礎,利用CP/MAS13C NMR光譜,在有序的C4區域(δ 86~92),用于模擬模型的包括來自結晶纖維素I的三種信號的Lorentzian譜線:纖維素 Iα(δ 89.6)、纖維素 Iα+β(δ 88.8)和纖維素Iβ(δ 88.0);在無序的區域(δ 80~86)是由非晶態纖維素引起的四種信號的Gaussian譜線:次晶纖維素(δ 88.5)、可及基原纖表面(δ 83.4與 δ 84.3)和不可及基原纖表面(δ 84.0),利用混合Lorentzian和Gaussian函數模型(如圖5)擬合技術就能準確分析纖維素不同結晶結構所占比例[13],再根據經驗公式q= ( 4n-4 ) / n2,計算橫向基原纖尺寸及基原纖聚集束尺寸[15](結果見表5)。
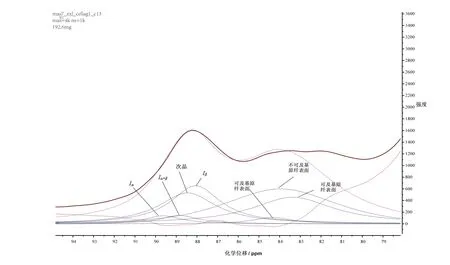
圖5 纖維素分子的CP/MAS 13C NMR光譜信號峰C4區的擬合結果Fig.5 Spectral fi tting of the C4 regions in CP/MAS 13C NMR spectrum
表5列出了幾種煙草纖維素樣品的CP/MAS13C NMR光譜C4區擬合得到的結晶和非晶區的比例及基原纖結構和基原纖聚集束的尺寸。由結晶區幾種晶型的擬合結果可進一步計算出結晶度,結果與由面積計算的結果基本一致(如表4示),一方面證實了在微原纖內部次晶纖維素的存在,另一方面也說明擬合結果的可靠性。
根據經驗方程計算的基原纖結構和基原纖聚集束的尺寸[15]表明,煙草纖維素的基原纖尺寸在3.0-5.0nm之間,與結晶度之間有明顯的相關性,基原纖聚集束的尺寸一般小于13nm。這些方法和數據可以用來進一步研究煙草加工和改性過程中纖維素晶體內部結構的變化及其對品質(如潤脹性能等)的影響,從而為煙草纖維素的改性提供理論依據。一般來說,在纖維素酸解過程中,由于半纖維素和木質素的脫除,可及基原纖表面增加,基原纖聚集束的尺寸會有所下降[19]。

表5 不同煙草纖維素樣品C4區光譜擬合的定量信息Tab.5 Quanti fi cation results from spectral fi tting of tobacco cellulose C4-region

續表5
3 結論
煙草纖維素晶型以Iβ晶型為主要晶體形式,煙草纖維素的結晶度在50%左右。煙草纖維素基原纖尺寸為3.0-5.0nm,基原纖聚集束的尺寸為6-13nm。本文采用13C NMR技術對煙草纖維素結構進行了初步探索,對于煙葉改性、酶解過程中纖維素結構水平上的變化(結晶度、微原纖、基原纖及纖維素鏈等發生結構的變化)還有待于進一步深入的研究。此項研究可以為纖維素改性、煙草加工以及低次煙葉和煙梗的處理等方面提供重要的研究基礎。
[1]任曉紅,陳剛,馬海燕,等.烤煙細胞壁物質對煙葉質量影響研究[J].中國農學通報2010,26(4): 113-116.
[2]周順, 徐迎波, 王程輝, 等.比較研究纖維素、果膠、淀粉的燃燒行為和機理[J].中國煙草學報, 2011, 17(5): 1-9.
[3]Henniges U, Kostic M, Borgards A, et al.Dissolution Behavior of Different Celluloses [J].Biomacromolecules 2011, 12: 870-879.
[4]Bokelman GH, Ryan WS Jr.Aanlyses of Bright and Burley tobacco laminae and stems[J].Beitr.Tabakforsch.Int.1985, 13:29-36.
[5]YC/T 347-2010 煙草及煙草制品 中性洗滌纖維、酸性洗滌纖維、酸洗木質素的測定 洗滌劑法.
[6]楊斌,殷引,張浩博,等.洗滌劑法測定煙草及煙草制品中中性洗滌纖維、酸性洗滌纖維、酸洗木質素的研究[J].中國煙草學報, 2012, 18(3): 10-15.
[7]Husson E, Buchoux S, Avondo C, et al.Enzymatic hydrolysis of ionic liquid-pretreated celluloses: Contribution of CP-MAS 13C NMR and SEM[J].Bioresource Technology, 2011, 102: 7335-7342.
[8]Holtman KM, Chen N, Chapopell MA, et al.Chemical structure and heterogeneity differences of two lignins from loblolly pine as investigated by advanced solid-state NMR spectroscopy [J].J Agric Food Chem, 2010, 58: 9882-9892.
[9]Mori T, Chikayama E, Tsuboi Y, et al.Exploring the conformational space of amorphous cellulose using NMR chemical shifts [J].Carbohydr Polym, 2013, 93: 122-128.
[10]Zuckerstatter G, Schild G, Wollbold, P, Rode, T, Weber HK, Sixta H (2009).The elucidation of cellulose supramolecular structure by 13C CP-MAS NMR[J].Lenzinger Berichte, 87, 38-46.
[11]Hall RA, Wooten JB.Quantitative Analysis of cellulose in tobacco by CP/MAS 13C NMR [J].J Agric Food Chem, 1998, 46: 1423-1427.
[12]Wickholm K, Larsson PT, Iversen T.Assignment of noncrystalline forms in cellulose by CP/MAS 13C NMR spectroscopy[J].Carbohydr Res,1998, 312: 123-129.
[13]Hult E, Liitia T, Maunu SL, et al.A CP/MAS 13C-NMR study of cellulose structure on the surface of re fi ned kraft pulp fi bers [J].Carbohydr Polym,2002, 49: 231-234.
[14]鄭明霞, 李來慶, 鄭明月, 等.堿處理對玉米秸稈纖維素結構的影響[J].環境科學與技術, 2012, 35: 27-31.
[15]肖青,萬金泉,王艷.CP/MAS 13C NMR技術對木漿纖維微觀結構的研究[J].化學學報, 2009, 67: 2629-2634.
[16]萬金泉,肖青,王艷.固體核磁共振和原子力顯微鏡分析不同半纖維素含量植物纖維的微觀結構[J].分析化學,2010, 38: 347-351.
[17]馬曉娟, 黃六蓮, 陳禮輝, 等.纖維素結晶度的測定方法[J].造紙科學與技術, 2012, 31, 75-78.
[18]Rondeau-Mouro C, Bizot H, Bertrand D.Chemometric analyses of the 1H-13C cross-polarization build-up of celluloses NMR spectra: A novel approach for characterizing the cellulose crystallites[J].Carbohydr Polym, 2011, 84, 539-549.
[19]Chunilall V, Bush T, Larsson PT, et al.A CP/MAS 13C NMR study of cellulose fi bril aggregation in eucalyptus dissolving pulps during drying and the correlation between aggregate dimensions and chemical reactivity[J].Holzforschung 2010, 64, 693-698.
Analysis of tobacco cellulose structure by CP / MAS13C NMR spectroscopy
FENG Guanglin1, TAN Lanlan1, ZHU Xiaolan2, GAO Yun2, DAI Ya1
1 Technology Center, China Tobacco Chuanyu Industrial Corporation, Chengdu 610066, China;2 Centre for Tobacco and Health Research, University of Science and Technology of China, Hefei 230052, China
A method utilizing CP / MAS13C NMR spectra was developed for analyzing tobacco cellulose structure.The NMR spectra in combination with spectral fitting were analyzed and structure parameters such as cellulose crystallinity, allomorph composition and lateral dimensions for cellulose elementary fi brils and micro fi brils were determined.Results showed that the main allomorph composition in tobacco cellulose was Iβ.The cellulose crystallinity calculated by spectral fi tting was about 50%.The lateral dimensions for cellulose elementary fi brils and micro fi brils were in the range of 3.0-5.0nm and 6.0-13.0nm, respectively.This study could provide theoretical basis for improving cellulose structure, and stem and leaf tobacco processing.
tobacco; cellulose; CP/MAS; NMR spectroscopy; crystalline structure
馮廣林,譚蘭蘭,朱曉蘭,等.CP/MAS13C NMR光譜法分析煙草纖維素的結構[J].中國煙草學報,2015,21(5 )
川渝中煙工業公司科技基金項目(合同號:JL/CYZY G SJ003-04)
馮廣林(1970—),碩士,高級工程師,主要從事土壤化學及煙草化學研究,Email:fengguanglin1973@yahoo.com.cn
戴亞,教授,Email: dycy@263.net
2014-06-14
:FENG Guanglin, TAN Lanlan, ZHU Xiaolan, et al.Analysis of tobacco cellulose structure by CP / MAS13C NMR spectroscopy[J].Acta Tabacaria Sinica, 2015,21(5)
猜你喜歡
奧秘(創新大賽)(2023年3期)2023-05-06 01:48:20
中國煙草學報(2019年5期)2019-11-14 07:54:12
首都公共衛生(2019年5期)2019-05-21 01:08:34
浙江中西醫結合雜志(2017年2期)2017-01-12 18:23:59
新聞傳播(2016年3期)2016-07-12 12:55:34
當代化工研究(2016年9期)2016-03-20 16:22:08
自動化博覽(2014年6期)2014-02-28 22:32:15
聲屏世界(2014年6期)2014-02-28 15:18:09
西南學林(2013年2期)2013-11-12 12:58:54
中國煙草學報(2012年5期)2012-04-12 06:21:18
