肥厚型心肌病致病基因型與臨床表現的關系及基因篩查在肥厚型心肌病篩查及疾病鑒別診斷中的作用*
2015-12-15 06:47:48崔宏麗王東馮新星鄒玉寶王怡璐王繼征惠汝太宋雷趙鵬
中國循環雜志 2015年2期
崔宏麗,王東,馮新星,鄒玉寶,王怡璐,王繼征,惠汝太,宋雷,趙鵬
肥厚型心肌病致病基因型與臨床表現的關系及基因篩查在肥厚型心肌病篩查及疾病鑒別診斷中的作用*
崔宏麗,王東,馮新星,鄒玉寶,王怡璐,王繼征,惠汝太,宋雷,趙鵬
目的:分析肥厚型心肌病(HCM) 致病基因型與臨床表現的關系及基因篩查在HCM篩查及疾病鑒別診斷中的作用。
方法:選擇一個HCM家系共14人,多重靶向測序技術對先證者的26個已知最常見的HCM致病基因進行全外顯子捕獲測序,用Sanger測序對發現的突變進行驗證并對其他家系成員及307名健康對照進行該突變位點的篩查,分析其基因型與臨床表型的特點。
結果:先證者及其子攜帶MYH7基因c.2146 G>A(Gly716Arg) 突變,該突變位于MYH7基因19號外顯子,導致第716位氨基酸殘基由Gly變為Arg,其他25個基因未發現突變。Sanger測序驗證后對其他家系成員進行突變篩查,其他家庭成員及對照組未發現該突變,該突變與HCM在該家系中共分離。先證者攜帶的致病突變為從頭突變,并遺傳給其子。先證者臨床表現為發病早(14歲)、勞力性呼吸困難、胸痛、心悸、心力衰竭,其子出生時即發現心肌肥厚。其父雖然室間隔肥厚(15 mm),但結合其年輕時曾為運動員的經歷及遺傳篩查的陰性結果,可基本排除其為HCM患者,考慮為生理性肥厚。
結論:該家系HCM由MYH7基因從頭突變p.Gly716Arg導致,該突變臨床發病早,癥狀較重,預后較差,為惡性突變。基因篩查在HCM家系篩查及疾病鑒別診斷中有重要意義。
肥厚型心肌病;MYH7基因;Gly716Arg;鑒別診斷
Methods: A HCM pedigree including 14 family members were studied. There were 26 known common HCM related genes were comprehensively screened for mutations in the proband with targeted resequencing. The mutation was identified by bi-directional Sanger sequencing, the specific mutation was examined in all family member and 307 healthy subjects. The relationship between the genotype and the phenotype was studied in this pedigree.
Results: Genetic testing identified a missense mutation, c. 2146 G>A (Gly716Arg) in 19thexon of β-myosin heavy chain gene (MYH7) in proband and her son. No other mutation was found in the rest of 25 genes. No same mutation was found in other family member and 307 healthy subjects. The proband carries a mutation causing HCM, it is a de novo mutation, and the mutation has been transmitted to her son. The proband had the early onset at 14
years of age, suffering from exertional dyspnea, chest pain, palpitation and heart failure; her son expressed HCM at new born. Although proband’s father having ventricular septal hypertrophy at 15mm, taking his athlete career and negative result of genetic screening, he could be basically excluded for HCM and considered as physiological cardiac hypertrophy.
Conclusion: A de novo mutation, Gly716Arg in MYH7 gene caused HCM in a specific Chinese pedigree. This mutation was malignant with early onset, severe symptoms and poor prognosis. Genetic screening is important for differential diagnosis of HCM.
(Chinese Circulation Journal, 2015,30:149.)
肥厚型心肌病(HCM)是最常見的單基因遺傳性心血管病,發病率為1/500[1]。該疾病臨床表現多樣,從無癥狀到呼吸困難、暈厥、心律失常、心力衰竭不盡相同,最惡劣的并發癥為心原性猝死(SCD),發生率約為0.8%[2]。HCM患者由于室間隔異常增厚使得左心室收縮時二尖瓣葉前移與室間隔的貼靠造成左心室流出道狹窄,射血量減少,導致活動時胸悶、暈厥等臨床癥狀,嚴重者可發生SCD[3],發生機制已證實多是由于室性心動過速或心室顫動所引起[4]。HCM為35歲以下青年人和運動員發生猝死的最主要原因, 給家庭和社會造成了重大損失[5]。大多數HCM患者為編碼心肌肌小節蛋白及其相關蛋白的基因突變引起的單基因常染色體顯性遺傳病[6]。最新的美國心臟病學會基金會/美國心臟協會(ACCF/ AHA)HCM診治指南推薦對HCM患者進行致病基因檢測,遺傳檢測有助于疾病確診和家系成員中患者的早期診斷[7]。但HCM有很強的臨床和遺傳異質性,目前尚缺少足夠的基因型—表型關聯,導致遺傳檢測在HCM預后判斷中的作用有限[2]。因此,通過家系分析,建立致病突變與臨床表型聯系,對于揭示HCM的遺傳特點、判斷預后、指導治療具有非常重要的意義。在本研究中,我們應用靶向重測序技術在一個HCM家系中對26個已知的最常見HCM致病基因進行了全面篩查,明確了該家系的致病基因和致病突變,分析了該致病突變導致的HCM的臨床特點,并且探討了基因篩查對于HCM家系成員篩查及疾病鑒別診斷的意義。
1 資料與方法
研究對象:2010-01 至2011-10收集的一個HCM家系共14人。其中男性8例,女性6例,年齡4~68歲,平均年齡(39.1±18.6)歲。先證者一位,女性,32歲。HCM診斷標準:成人超聲心動圖檢查室間隔或心室壁厚度≥13 mm,排除長期難以控制的高血壓、主動脈瓣狹窄等引起繼發性心肌肥厚的心血管系統疾病和全身性疾病。選取307例心電圖和心動超聲圖檢查未示異常的健康志愿者作為對照,其中男性194例,女性113例,平均年齡(53.1±10.7)歲。本研究經阜外心血管病醫院倫理委員會批準實施。
采集HCM家系的臨床資料:體格檢查包括身高、體重、血壓、心率等;心電圖、二維及多普勒超聲心動圖檢查。
基因組DNA提取:采集受調查者外周靜脈血4 ml,四乙酸二氨基乙烷(EDTA)抗凝。利用血液基因組提取試劑盒(貨號DP319,天根生化科技有限公司,北京) 提取基因組DNA。
先證者基因測序:檢測的26個致病基因包括:MYH7, MYBPC3, TNNT2, TNNI3, MYL2, MYL3,TPM1, ACTC1, MYH6, TNNC1, TTN, ACTN2,TCAP, VCL, ANKRD1, CAV3, CSPR3, LDB3,MYOZ2, NEXN, JPH2, PLN, CASQ2, CALR3,PRKAG2 和LAMP2。利用高通量測序法對先證者的26個致病基因的編碼外顯子和上下游5 bp內含子序列進行靶向測序,基因組DNA經超聲破碎為250 bp左右長度片段,利用AMPureXP磁珠(Agencourt,Beckman Coulter, CA, USA)富集200~300 bp范圍內的片段。連接通用接頭后,聚合酶鏈式反應(PCR)擴增8個循環,利用定制的Agilent液相捕獲文庫(Agilent Technologies, Santa Clara, CA, USA)富集目標區域。富集后的基因組片段經過10個循環括增后,Illumina GA IIx (Illumina Inc, CA, USA)進行雙端測序,每端讀長120 bp。測序數據利用PICARD去除重復后,利用CLC Genomics Workbench (CLC-bio,Aarhus, Denmark)與人類基因組參考序列(GRCh37/ hg19)進行比對,分析26個致病基因目標區域的
覆蓋度和變異。變異須滿足所在位點測序深度≥25×,并且變異堿基所占比例≥20%。
家系及健康對照的特定基因測序:應用Oligo 6.0軟件進行引物設計,由生工生物工程(上海)股份有限公司合成。因MYH7第19號外顯子較短,故設計引物時將其與第20號外顯子合并在一起。PCR擴增MYH7基因第(19+20)號外顯子,引物上游序列:5' CAA AGC CAG GAT CAG AAC CCA G 3';下游序列:5' GGA GTC AAT GGA AAA GAG ATG T 3'。PCR擴增體系20 μl:含基因組DNA 20 ng,上下游引物各0.8 pmol,2×Taq MasterMix(北京康為世紀生物科技有限公司)10 μl。擴增條件:預變性,95℃ 10 min;變性95℃ 30 s,退火52℃30 s,延伸72℃ 40 s,共35個循環;最后延伸72℃,10 min。(PCR儀為FACS Calibur 、美國 BD Biosciences公司產品)
PCR產物純化后,按雙脫氧末端終止法(Sanger法)進行測序( 美國Applied Biosystems公司 3730XL測序儀),測序引物與PCR引物相同。第(19+20)號外顯子的目的片段大小約514 bp。
生物信息分析:變異在人群中的頻率通過檢索Exome Sequencing Project 數據庫(ESP,http:// evs.gs.washington.edu/EVS)和1 000 Genomes數據庫(http://www.1000genomes.org)確定。變異致病性通過PolyPhen-HCM(www.http://genetics.bwh.harvard.edu/ hcm)預測。
2 結果
HCM家系(圖1)的臨床及遺傳學特點:先證者及其子攜帶Gly716Arg突變,均呈現臨床肥厚型心肌病表現(圖2)。①先證者(Ⅲ-6),女性,32歲,14歲時因發熱入院,心電圖提示ST-T改變。1997年確診為HCM,主要癥狀為勞力性呼吸困難、胸痛,當時超聲檢查示:非對稱性肥厚型心肌病(非梗阻型),室間隔中下段明顯肥厚(20 mm),左心室舒張末期內徑36 mm,左心室射血分數67%,左心房內徑30 mm。2007年分娩時出現心力衰竭,之后出現心悸、頭暈等癥狀,無黑矇、暈厥。2014年復查超聲:室間隔厚25 mm,左心室舒張末期內徑44 mm,左心室射血分數54%,左心房內徑40 mm(圖2A)。②先證者之子(Ⅳ-3),4歲,出生時心肌即有肥厚(出生時臨床資料缺失)。兩歲時超聲檢查示:室間隔明顯增厚(15 mm),左心室舒張末期內徑29 mm,左心室射血分數78%,左心房內徑25 mm,左冠狀動脈內徑增寬。安靜心電圖:室內傳導阻滯,T波倒置(下壁),ST-T異常(側壁),異常Q波。動態心電圖:竇性心律不齊,竇房結內游走伴不齊,異常Q波,ST-T改變,心率變異性:正常R-R間期標準差>50 ms。2014年復查超聲:室間隔厚16 mm,左心室舒張末期內徑32 mm,左心室射血分數75%,左心房內徑26 mm(圖2B)。③先證者之父(Ⅱ-1),無癥狀,超聲檢查示:室間隔厚15 mm,左心室舒張末期內徑40 mm,左心室射血分數68%,左心房內徑25 mm,超聲心動圖正常(圖2C)。基因篩查未發現突變。結合其年輕時曾為運動員的經歷,考慮為生理性肥厚,并無臨床癥狀,無需治療。④ 家系成員中的Ⅱ-3有不典型性胸痛、心悸等臨床表現,Ⅱ-5有不典型胸痛的臨床表現,但是兩者的超聲檢查均未發現心肌肥厚,心電圖檢查亦無異常,基因篩查也沒有檢測到突變。其余家系成員的臨床表現和各項檢查均無異常。
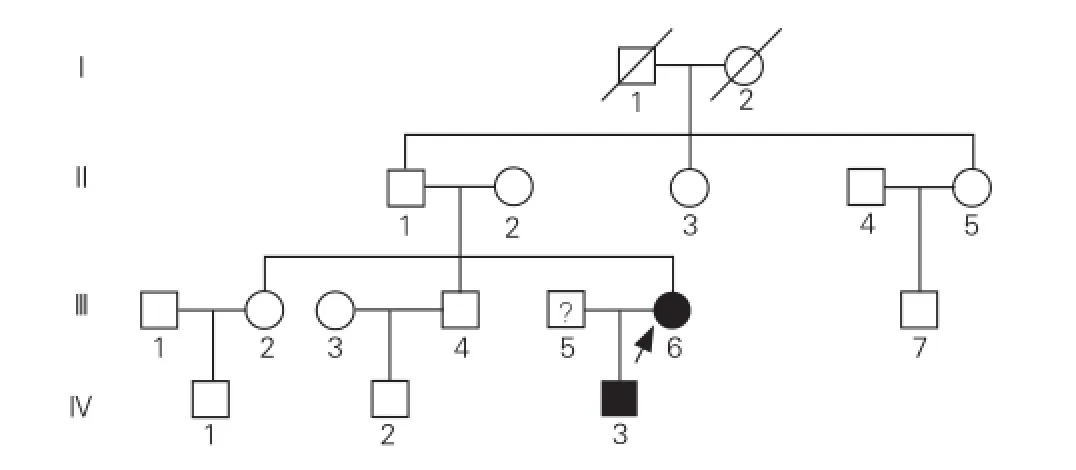
圖1 肥厚型心肌病家系圖譜
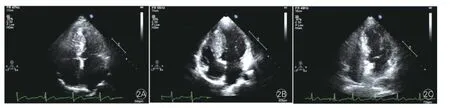
圖2 先證者及其父、其子的超聲心動圖
HCM家系基因測序結果:先證者及其子攜帶MYH7基因c.2146 G>A(Gly716Arg) 突變(圖3A)。該突變位于MYH7基因19號外顯子,導致第716位氨基酸殘基由Gly變為Arg,其他25個基因未發現突變。Sanger測序驗證后對其他家系成員進行突變篩查(圖1、3),其他家庭成員及對照組未發現該突變,同源性比對發現MYH7基因Pro716氨基酸殘基在不同物種間高度保守(圖3B),該突變與HCM在該家系中共分離。
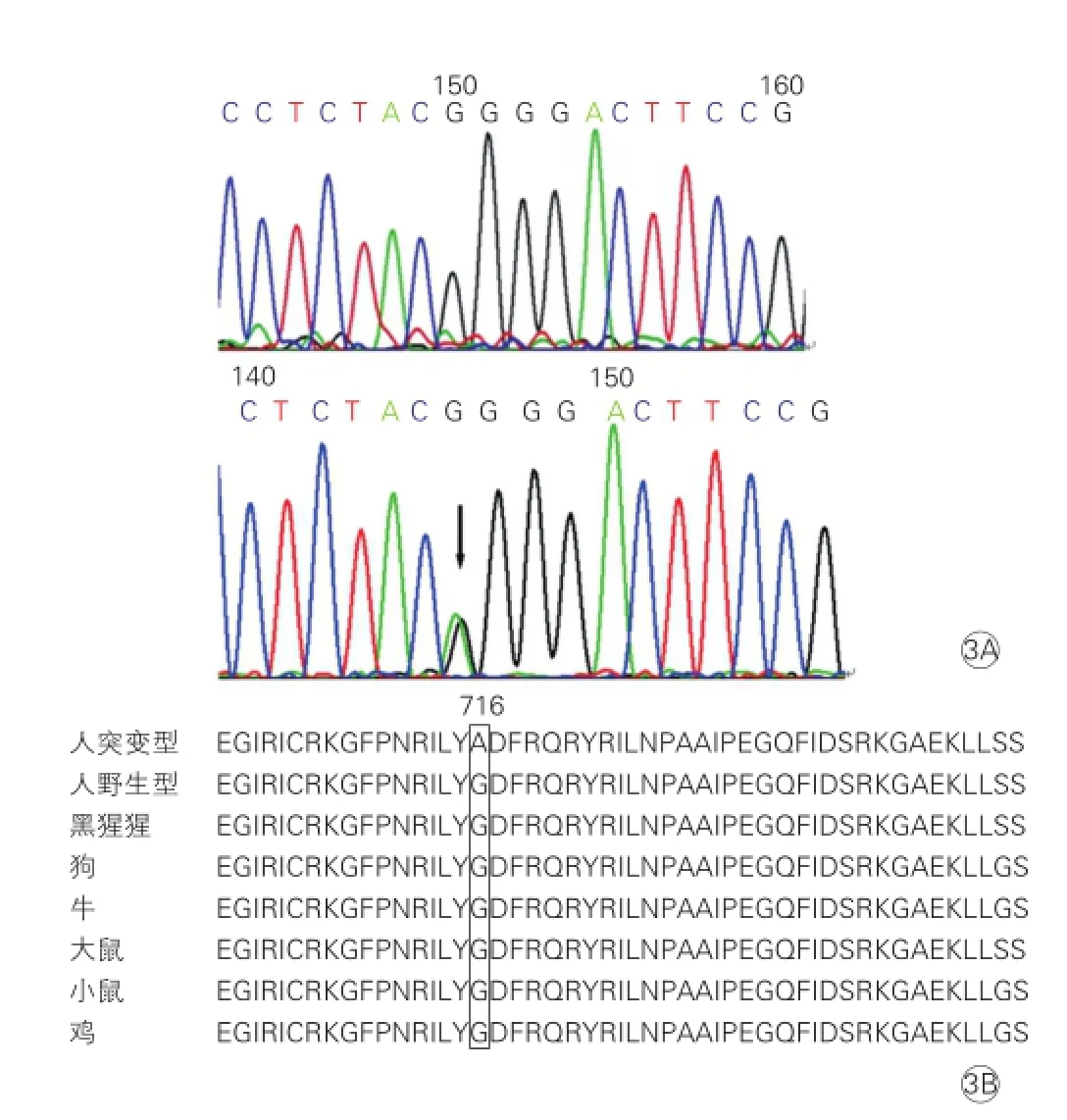
圖3 MYH7基因Gly716Arg突變測序圖及物種間保守性分析
3 討論
MYH7的突變所引起的HCM占所有病例的35%~50%,它是第一個被確定的也是最常見的HCM致病基因之一[8]。它編碼的蛋白是含1935個氨基酸的肌球蛋白重鏈-β-MHC。研究發現,發生在MYH7基因上的突變并不是均勻普遍的分布的,也就是說,有聚集性,好發部位主要是β-MHC頭部的5個區域,而位于其他部位的突變則沒有那么常見[9,10]。
MYH7基因Gly716Arg變異在ESP和1000 Genomes多態數據庫中均無報道。同源性比對發現MYH7基因Pro716氨基酸殘基在不同物種間高度保守。MYH7基因Gly716Arg突變位于19號外顯子,19號外顯子編碼對應β-MHC頭部的靠近鉸鏈區的2個半胱氨酸連接區,發生在此區的突變還包括Arg719Trp、Arg719Gln、Arg723Gly和Gly741Arg。該部位為β-MHC的重要功能區,發生突變可以增加肌球蛋白S1的ATP酶活性,推斷其致病機制為阻礙了肌球蛋白的構象改變或改變了肌球蛋白與肌動蛋白及其他分子的相互作用,引起嚴重的臨床表現[11]。
Gly716Arg突變外顯率高,攜帶者發病早、進行性心力衰竭、猝死率高,為惡性突變。Rai等[11]的研究發現此突變攜帶者具有嚴重的不對稱性室間隔肥厚(31 mm),先證者在21歲時發生猝死。在Anan等[12]做的一項研究中,攜帶此突變的家系成員發病早,進行性左心室壁變薄、收縮功能減退,頑固性心力衰竭。Arg719Trp、Arg719Gln、Arg723Gly和Gly741Arg突變引起的表型也有相同的惡性臨床表現[12-17]。
HCM一直被認為是遵循常染色體顯性遺傳模式,患者的一級親屬均有50%的可能攜帶有與患者相同的致病基因,并存在發病的潛在風險[6]。傳統的對于HCM的診斷以及猝死危險的評估首先都是基于出現左心室肥厚,但有很大局限性。而基因篩查能夠為篩選出家系中有發病可能的親屬提供較為可靠的證據并實現HCM與擬表型疾病(如Fabry病、LEOPARD綜合征)的鑒別[18,19]。它還能解決HCM與運動員型心臟以及高血壓心肌肥厚的鑒別難題[20,21]。
本研究中先證者之父超聲示心肌肥厚(室間隔厚15 mm),符合HCM診斷標準,若不做基因篩查,則極可能依據超聲及家族史而診斷為HCM。但先證者及其子攜帶的致病突變并未在他身上檢測到,再結合其年輕時長期從事高強度運動的經歷,綜合考慮其心肌肥厚為生理性肥厚,現無臨床表現故無需進行治療,僅進行隨訪觀察即可。
目前國內還沒有關于Gly716Arg突變的報道,本研究發現的漢族家族性HCM中兩例此突變攜帶者皆發病早(先證者14歲,其子出生即發現心肌肥厚),先證者心肌中度肥厚、勞力性呼吸困難,年紀尚輕就發生心力衰竭,與國外報道的惡性表現基本相符,應對攜帶者進行積極的觀察和治療,預防猝死的發生。
值得強調的是只有50%的先證者在基因篩查中被檢測到,說明仍然有很多與HCM相關的基因未被識別而未能被引入到候選基因范疇[22],并且有時先證者攜帶有兩個致病突變但僅查出一個,或者出現了從頭突變,都會影響到基因檢測的結果[23]。仍需要大規模多中心臨床研究為同時基于基因檢測和臨床表型的HCM治療和預防策略的制定提供充分的依據。
本研究中,先證者攜帶的突變為從頭突變,即先證者的父母并不攜帶突變且無HCM的臨床表現。它的發生率是非常低的,徐榮等[24]于2003年報道了中國首例從頭突變。從頭突變進一步證明了MYH7基因為HCM的責任基因。本研究中先證者之子也遺傳了該突變,由此看來,散發性肥厚型心肌病也應警惕其將突變傳遞給下一代的風險。
這是國內較早發現的MYH7基因Gly716Arg突變,屬惡性突變。本研究進一步說明了基因篩查對于HCM家系篩查及疾病鑒別診斷的重要意義以及應警惕散發性肥厚型心肌病將其突變傳遞給下一代的風險。
[1] Maron BJ. Hypertrophic cardiomyopathy: a systematic review. J Am Meu Assc, 2002, 287: 1308-1320.
[2] Maron BJ, Olivotto I, Spirito P, et al. Epidemiology of hypertrophic cardiomyopathy related death: revisited in a large non-referral-based patient population. Circulation, 2000, 102: 858-864.
[3] 侯翠紅, 喬樹賓, 楚建民, 等. 肥厚型梗阻性心肌病行左心室流出道疏通術與經皮室間隔化學消融術治療的遠期療效分析. 中國循環雜志, 2010, 25: 38-40.
[4] 崔彬, 許建屏, 王巍. 肥厚型梗阻性心肌病圍手術期心律失常特點及治療策略. 中國循環雜志, 2011, 26: 129-132.
[5] Maron BJ, Maron MS, Semsarian C. Genetics of hypertrophic cardiomyopathy after 20 years: clinical perspectives. J Am Coll Cardiol, 2012, 60: 705-715.
[6] Elliott P, McKenna WJ. Hypertrophic cardiomyopathy. Lancet, 2004, 363: 1881-1891.
[7] Gersh BJ, Maron BJ, Bonow RO, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation, 2011, 124: e783-831.
[8] Tanjore R, RangaRaju A, Vadapalli S, et al. Genetic variations of β-MYH7 in hypertrophic cardiomyopathy and dilated cardiomyopathy. Indian J Hum Genet, 2010, 16: 67-71.
[9] Richard P, Charron P, Carrier L, et al. Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for amolecular diagnosis strategy. Circulation, 2003, 107: 2227-2232.
[10] Yu B, Sawyer NA, Caramins M, et al. Denaturing high performance liquid chromatography: high throughput mutation screening in familial hypertrophic cardiomyopathy and SNP genotyping in motor neurone disease. J Clin Pathol, 2005, 58: 479-485.
[11] Rai TS, Ahmad S, Bahl A, et al. Early expression of a malignant phenotype of familial hypertrophic cardiomyopathy associated with a Gly716Arg myosin heavy chain mutation in a Korean family. Am J Cardiol, 1998, 82: 1509-1513.
[12] Anan R, Greve G, Thierfelder L, et al. Prognostic implications of novel beta cardiac myosin heavy chain gene mutations that cause familial hypertrophic cardiomyopathy. J Clin Invest, 1994, 93: 280-285.
[13] 宋雷, 黃曉紅, 惠汝太, 等. 心肌球蛋白重鏈基因Arg719Gln突變與家族性肥厚型心肌病. 中華心血管病雜志, 2001, 29: 348-352.
[14] Enjuto M, Francino A, Navarro-López F, et al. Malignant hypertrophic cardiomyopathy caused by the Arg723Gly mutation in beta-myosin heavy chain gene. J Mol Cell Cardiol, 2000, 32: 2307-2313.
[15] 鄭冬冬, 楊俊華, 董寧征, 等. 中國漢族家族性肥厚型心肌病人群MYH7基因Arg723Gly突變分析. 中華心血管病雜志, 2006, 34: 208-211.
[16] D?hlemann C, Hebe J, Meitinger T, et al. Apical hypertrophic cardiomyopathy due to a de novo mutation Arg719Trp of the betamyosin heavy chain gene and cardiac arrest in childhood. A case report and family study. Z Kardiol, 2000, 89: 612-619.
[17] Miller G, Colegrave M, Peckham M. N232S, G741R and D778G beta-cardiac myosin mutants, implicated in familial hypertrophic cardiomyopathy, do not disrupt myofibrillar organisation in cultured myotubes. FEBS Lett, 2000, 486: 325-327.
[18] O'Mahony C, Lambiase PD, Quarta G, et al. The long-term survival and the risks and benefits of implantable cardioverter defibrillators in patients with hypertrophyic cardiomyopathy. Heart, 2012, 98: 116-125.
[19] Weidemann F, Niemann M, Breunig F, et al. Long-term effects of enzyme replacement therapy on Fabry cardiomyopathy Evidence for a better outcome with early treatment. Circulation, 2009, 119: 524-529.
[20] Patel MR, Cecchi F, Cizmarik M, et al. Cardiovascular events in patients with Fabry disease natural history data from the Fabry registry. J Am Coll Cardiol, 2011, 9: 1093-1099.
[21] Andersen PS, Havndrup O, Hougs L, et al. Diagnostic yield, interpretation, and clinical utility of mutation screening of sarcomere encoding genes in Danish hypertrophic cardiomyopathy patients and relatives. Hum Mutat, 2009, 30: 363-370.
[22] Ackerman MJ, Priori SG, Willems S, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies. Heart Rhythm, 2011, 8: 1308-1339.
[23] Erdmann J, Daehmlow S, Wischke S, et al. Mutation spectrum in a large cohort of unrelated consecutive patients with hypertrophic cardiomyopathy. Clin Genet, 2003, 64 : 339-349.
[24] 徐榮, 王繼征, 鄒玉寶, 等. 心肌肌鈣蛋白I基因Arg170Gln突變—de novo突變與肥厚型心肌病. 中華醫學雜志, 2003, 83: 1634.
Relationship Between the Genotype Causing Hypertrophic Cardiomyopathy With its Clinical Presentation and Differential Diagnosis
CUI Hong-li, WANG Dong, FENG Xin-xing, ZOU Yu-bao, WANG Yi-lu, WANG Ji-zheng, HUI Ru-tai, SONG Lei, ZHAO Peng. Department of Pathology, Affliated Hospital, Qingdao University Medical College, Qingdao (266003), Shandong, China
Co-corresponding Authors: SONG Lei, Email: lsongqd@yahoo.com and ZHAO Peng, Email: zhpeng17@hotmail.com
Objective: To identify the mutation gene for Chinese Han population with hypertrophic cardiomyopathy (HCM), to analyze the relationship between the genotype and phenotype, and to study the effect of genetic screening in HCM pedigrees for differential diagnosis of HCM.
Hypertrophic cardiomyopathy; MYH7gene; Gly716Arg; Differential diagnosis
2014-03-12)
(編輯: 曹洪紅)
青島市產學研合作引導計劃(應用基礎研究)(13-1-4-141-jch)
266003 山東省青島市,青島大學醫學院附屬醫院 病理科(崔宏麗、趙鵬);中國醫學科學院 北京協和醫學院 國家心血管病中心阜外心血管病醫院 心血管疾病國家重點實驗室(王東、馮新星、鄒玉寶、王怡璐、王繼征、惠汝太、宋雷)
崔宏麗 碩士研究生 研究方向為肥厚型心肌病與心原性猝死 Email:cui1998322@163.com 通訊作者:宋雷 Email:lsongqd@yahoo.com趙鵬 Email:zhpeng17@hotmail.com
R541
A
1000-3614(2015)02-0149-05
10.3969/j.issn.1000-3614.2015. 02.014
