二甲雙胍降低2型糖尿病大鼠主動脈磷酸化絲裂原活化蛋白激酶的蛋白表達
2015-12-16 01:22:06陳麗莉范國洽韓蕊石志平王濤張潔王雪周亞茹
中國循環雜志 2015年5期
陳麗莉,范國洽,韓蕊,石志平,王濤,張潔,王雪,周亞茹
二甲雙胍降低2型糖尿病大鼠主動脈磷酸化絲裂原活化蛋白激酶的蛋白表達
陳麗莉,范國洽,韓蕊,石志平,王濤,張潔,王雪,周亞茹
目的:探討p38絲裂原活化蛋白激酶( MAPK)與2型糖尿病大鼠主動脈病變的關系及二甲雙胍的干預作用。
方法:25只雄性Wistar大鼠隨機分為對照組和實驗組,實驗組采用高脂高糖飼料喂養聯合鏈脲佐菌素注射建立2型糖尿病大鼠模型。將成模糖尿病大鼠隨機分為2組:2型糖尿病空白對照組(2型糖尿病組,n=7)、2型糖尿病+二甲雙胍干預組(二甲雙胍組,n=8)。二甲雙胍組給予二甲雙胍200 mg/kg灌胃8周,于干預末行腹腔葡萄糖耐量試驗,次晨心尖取血并分離血清,全自動生化分析儀測定血糖、血脂等生化指標;放免法測定胰島素水平;采用酶聯免疫吸附方法測定血清細胞間黏附分子-1( ICAM-1), 血管細胞黏附分子-1(VCAM-1)和核因子-κB水平;取胸主動脈進行蘇木素伊紅(HE)染色,光鏡下觀察血管病理變化;免疫組化法檢測大鼠胸主動脈磷酸化p38 MAPK、核因子-κB、單核細胞趨化蛋白-1蛋白的表達。
結果:(1)HE染色可見2型糖尿病組大鼠主動脈管壁結構層次不清,內膜增厚,內皮細胞腫大變性,中膜平滑肌細胞排列紊亂,膠原纖維增生。(2)與對照組相比,2型糖尿病組大鼠空腹胰島素[(20.00 ± 5.91) mIU/L vs(11.34 ± 3.88)mIU/L]、核因子-κB [(170.22±21.53) μmol/L vs(78.68±12.15) μmol/L]、ICAM-1[(130.59±16.00) pg/ml vs(75.63±19.52)pg/ml]、VCAM-1 [(990.19±119.18) ng/ml vs(616.54±54.51) ng/ml]、總膽固醇 [(4.15±0.41) mmol/L vs(2.18±0. 93 ) mmol/L]、甘油三酯 [(1.83±0.40) mmol/L vs(0.81±0.27)mmol/L]、低密度脂蛋白膽固醇[(2.53±0.44) mmol/L vs (1.24±0.47)mmol/L]水平明顯升高(P<0.05),胰島素曲線下面積(30.78 ± 11.25)mIU·L-1·h vs(47.55 ± 5.23)mIU·L-1·h明顯降低(P<0.05),主動脈磷酸化p38 MAPK、核因子-κB、MCP-1蛋白表達水平升高(P<0.05),差異均有統計學意義。(3)實驗結束時二甲雙胍組大鼠空腹血糖、空腹胰島素、核因子-κB、單核細胞趨化蛋白-1、VCAM-1、甘油三酯、總膽固醇水平較2型糖尿病組明顯降低(P< 0.05);二甲雙胍組和2型糖尿病組間胰島素曲線下面積無明顯差異(P> 0.05)。二甲雙胍組大鼠主動脈磷酸化p38 MAPK、核因子-κB、單核細胞趨化蛋白-1蛋白表達水平也較二甲雙胍組明顯降低(P<0.05),差異有統計學意義。
結論:p38 MAPK信號通路的激活在糖尿病大血管病變發生發展中起重要作用,二甲雙胍通過降低p38 MAPK磷酸化水平、抑制炎癥反應、調節脂代謝而起到血管保護作用。
糖尿病;動脈粥樣硬化;p38絲裂原活化蛋白激酶;二甲雙胍
(Chinese Circulation Journal, 2015,30:487.)
研究顯示,動脈粥樣硬化的病理改變與絲裂原活化蛋白激酶(MAPK)的過度激活有關[1,2]。磷酸化p38 MAPK通過啟動核因子-κB,促進內皮細胞分泌血管細胞黏附分子-1(VCAM-1)、細胞間黏附分子-1(ICAM-1)表達,加劇中性粒細胞和單核細胞與內皮細胞黏附,誘發血栓形成和血管再狹窄,介導動脈粥樣硬化形成。
二甲雙胍除能夠有效降糖、降低胰島素水平及增加胰島素敏感性外,還有抗炎、抗氧化應激、改善血脂、改善血管內皮功能等作用[3-5]。本實驗通過高糖、高脂飲食聯合小劑量鏈脲佐菌素(STZ)制備2型糖尿病大鼠模型,繼而采用二甲雙胍進行干預治療,實驗結束后檢測各組大鼠血糖、血脂等生化指標,血清核因子-κB、VCAM-1、ICAM-1水平變化及胸主動脈中磷酸化的p38 MAPK蛋白表達水平,探討p38 MAPK與糖尿病動脈粥樣硬化的關系及二甲雙胍對2型糖尿病動脈粥樣硬化的影響及其機制。
1 材料與方法
實驗對象及分組:實驗時間為2012-11至2013-08,25只體重80 g左右的4周齡Wistar雄性大鼠(SPF級,購自河北醫科大學基礎醫學院動物實驗中心),采用數字表法隨機分為對照組(n=6)和實驗組(n=19),對照組采用標準飼料喂養;實驗組采用高脂高糖飼料喂養(含20%蔗糖、10%熟豬油、2.5%膽固醇、1%膽酸、66.5%標準飼料)。喂養4周后,實驗組大鼠隔夜空腹腹腔注射STZ(美國Sigma公司,30 mg/kg,用前以檸檬酸緩沖液配成1%的濃度),對照組僅注射等容積的檸檬酸緩沖液。于實驗的第6周即注射STZ 2周后,空腹8小時測血糖≥ 7.8 mmol/L者為糖尿病成模標準[6]。19只實驗大鼠中造模成功者15只。將成模實驗大鼠隨機分為2組:2型糖尿病空白對照組(2型糖尿病組, n=7)、2型糖尿病+二甲雙胍干預組(二甲雙胍組,n=8,二甲雙胍由中美上海施貴寶制藥有限公司提供)。二甲雙胍組給予二甲雙胍200 mg/kg[7]溶于飲用水中灌胃,每日1次,對照組和2型糖尿病組給予等量飲用水灌胃,每周測體重1次,共8周,整個實驗過程共持續14周。二甲雙胍組有1只大鼠在實驗過程中死亡(可能死于高血糖)。
觀察指標及檢測方法:二甲雙胍干預第8周末,行腹腔葡萄糖耐量試驗(禁食12 h,腹腔注射葡萄糖2 g/kg,在注射葡萄糖后0、30、60、90、120 min尾靜脈采血),采用近似梯形的計算公式計算葡萄糖曲線下面積和胰島素曲線下面積。次日麻醉下心尖取血,分離血清,檢測血清總膽固醇(TC)、甘油三酯(TG)、低密度脂蛋白膽固醇(LDL-C)、高密度脂蛋白膽固醇(HDL-C)、 核因子-κB、ICAM-1、VCAM-1水平。酶聯免疫吸附試劑盒購自bio-swamp公司。碘[125I]胰島素放射免疫分析藥盒購自北京北方生物技術研究所。
取約1 cm胸主動脈以4%多聚甲醛固定,進行蘇木素伊紅(HE)染色,光鏡下觀察血管病理變化;免疫組化法測定胸主動脈中磷酸化的p38 MAPK、核因子-κB、單核細胞趨化蛋白-1的蛋白表達水平。兔抗大鼠單核細胞趨化蛋白-1、核因子-κB抗體購自武漢博士德生物公司,兔抗大鼠p38 MAPK單克隆抗體購自上海bioworld公司。
統計學分析:采用SPSS16.0統計軟件進行分析,正態分布的數據以均數±標準差表示,非正態分布數據以中位數(最小值,最大值)表示。正態分布及方差齊性資料采用 t檢驗及單因素方差分析,組間兩兩比較采用 SNK-q 檢驗;不符合正態分布的資料,采用非參數檢驗。以P<0.05 為差異有統計學意義。
2 結果
實驗結束時大鼠血清生化指標比較(表 1):2型糖尿病組和二甲雙胍組大鼠空腹血糖水平較對照組明顯升高(P<0.05),二甲雙胍組空腹血糖較2型糖尿病組明顯下降 [(9.03±1.49)mmol/L vs(12.99±2.07)mmol/L,P<0.05];2型糖尿病組和二甲雙胍組葡萄糖曲線下面積水平明顯高于對照組(P<0.05),但前兩組組間葡萄糖曲線下面積比較無統計學意義(P>0.05)。2型糖尿病組大鼠空腹胰島素明顯高于對照組和二甲雙胍組(P<0.05),對照組胰島素曲線下面積水平明顯高于2型糖尿病組和二甲雙胍組(P<0.05),但2型糖尿病組和二甲雙胍組的胰島素曲線下面積水平比較差異無統計學意義(P>0.05)。2型糖尿病組和二甲雙胍組大鼠TG 、TC、LDL-C、ICAM-1、VCAM-1、核因子-κB水平均明顯高于對照組(P<0.05),二甲雙胍組TG 、TC、ICAM-1、VCAM-1、核因子-κB水平低于2型糖尿病組(P<0.05);與對照組相比,2型糖尿病組和二甲雙胍組HDL-C水平顯著降低(P<0.05)。
表1 實驗結束時三組大鼠血清生化指標
注:與對照組比*P<0.05;與2型糖尿病組比△P<0.05。#1只大鼠在實驗過程中死亡
?
三組大鼠病理形態學比較(圖1):對照組可見血管壁結構清晰,內膜光滑完整,平滑肌細胞排列整齊,內皮細胞結構完整。2型糖尿病組主動脈管壁結構層次不清,內膜增厚,中膜平滑肌細胞排列紊亂,膠原纖維增生血管壁彌漫性隆起,內膜明顯增厚,內膜下平滑肌細胞排列紊亂、增生,內有脂質沉積,膠原纖維和彈性纖維增多。二甲雙胍組血管內皮細胞基本完整,內膜偶有增厚,中膜平滑肌細胞排列基本整齊。
三組大鼠p38 MAPK、核因子-κB、單核細胞趨化蛋白-1蛋白表達的改變(圖2):胸主動脈p38 MAPK、核因子-κB、單核細胞趨化蛋白-1蛋白表達于血管內膜細胞胞質內,呈棕黃色顆粒沉著。免疫組織化學表達積分見表2。與對照組相比,2型糖尿病組主動脈磷酸化p38 MAPK、核因子-κB、單核細胞趨化蛋白-1表達水平升高,差異有統計學意義(P<0.05);與2型糖尿病組相比,二甲雙胍組主動脈磷酸化p38 MAPK、核因子-κB、單核細胞趨化蛋白-1表達水平降低(P<0.05)。
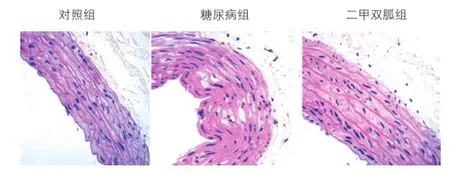
圖1 各組大鼠主動脈蘇木素伊紅染色(×200倍)
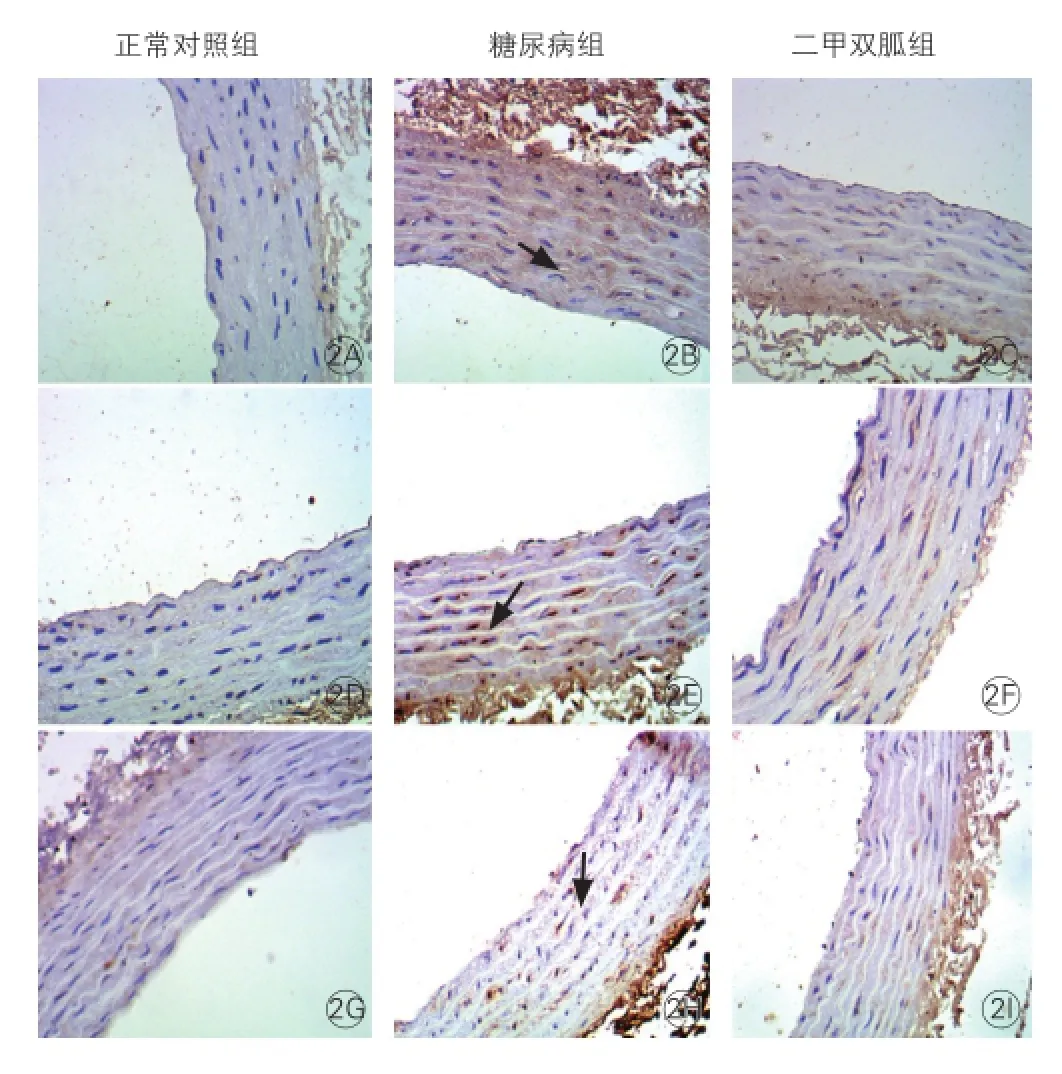
圖2 三組大鼠主動脈中單核細胞趨化蛋白-1、 核因子-κB、p38絲裂原活化蛋白激酶的表達(×200)
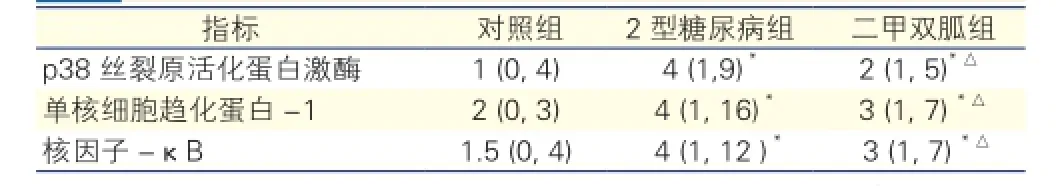
表2 三組大鼠免疫組化表達積分#
3 討論
糖尿病大血管病變的病理基礎為動脈粥樣硬化,它與單純的動脈粥樣硬化相比病變范圍大、程度重、發生早[8]。然而,糖尿病大血管病變的發病機理尚未完全闡明,目前認為,2型糖尿病大血管病變除與糖脂代謝紊亂有關外,尚與其他因素,如血管內皮功能損傷、炎癥和氧化應激、 MAPK介導的信號轉導通路激活有關[9,10]。 Yoon等[11]研究表明,晚期糖基化終末產物(AGE)可誘導血管平滑肌細胞增殖、形成活性氧簇,誘導核因子-κB、促MAPK信號轉導通路的激活,從而使炎癥因子的表達增加,血管內皮功能受損,導致糖尿病大血管病變的發生。上述研究提示,糖尿病大血管病變與炎癥因子和內皮功能障礙有關。MAPK介導的信號轉導通路是生物信號引起核反應的重要通路,在細胞生長、增殖、分化及凋亡的調節中起到至關重要的作用,動脈粥樣硬化的病理改變與MAPK的過度激活有關[1,2],其中p38 MAPK通路與內皮細胞損傷關系密切,是參與炎癥反應的細胞內的重要通路:非磷酸化的p38 MAPK通過磷酸化狀態,促進下游底物磷酸化來快速實現信號傳遞,啟動核因子-κB,促進內皮細胞分泌VCAM-1、ICAM-1,加劇中性粒細胞和單核細胞與內皮細胞黏附,誘發血栓形成和血管再狹窄,介導炎癥反應,參與動脈粥樣硬化形成。2型糖尿病患者存在的胰島素抵抗、高血糖和(或)高游離脂肪酸水平、以及炎性細胞因子等均能激活p38 MAPK信號通路[12]。本實驗中,2型糖尿病組大鼠胸主動脈HE染色可見主動脈發生了炎癥和損傷,另外,2型糖尿病組大鼠主動脈p38 MAPK蛋白表達水平及血清中核因子-κB、VCAM-1、ICAM-1水平明顯升高,說明p38 MAPK及炎癥因子參與了2型糖尿病大血管病變的發生、發展。
本實驗結果顯示,二甲雙胍組空腹血糖較2型糖尿病組明顯降低,但葡萄糖曲線下面積無統計學差異,可能與二甲雙胍主要通過減少肝糖輸出降低空腹血糖,但對餐后血糖作用較弱有關。胰島素曲線下面積在2型糖尿病組有明顯的降低,可能與糖尿病時高糖狀態對胰島β細胞的抑制狀態有關。同時,本實驗發現,相對于2型糖尿病組,二甲雙胍組可見到胰島素曲線下面積有增加的趨勢,可能與二甲雙胍抑制肝糖輸出、促進葡萄糖的攝取和利用,降低空腹血糖、改善葡萄糖負荷后血糖,一定程度改善胰島β細胞分泌功能有關。
現有研究認為,有效地控制血糖可延緩但不能防止2型糖尿病患者大血管并發癥的發生,這可能與2型糖尿病患者常合并血脂代謝異常、高血壓,存在氧化應激、凝血及纖溶系統紊亂等因素有關[13,14]。研究表明,二甲雙胍能改善血脂水平、抑制氧化應激和黏附分子的形成,能夠抗炎、改善血管內皮功能并激活纖溶系統,具有降糖以外的大血管保護作用[3-5]。本實驗證實,二甲雙胍組的血清TC、TG、核因子-κB、VCAM-1、ICAM-1水平較糖尿病組明顯降低,且胸主動脈組織炎細胞浸潤等情況明顯改善,進一步說明二甲雙胍除降糖外,還可起到改善血脂、抗炎、改善血管內皮功能等作用,從而改善糖尿病大血管病變。本研究還發現,與實驗組相比,二甲雙胍組p38 MAPK、核因子-κB、單核細胞趨化蛋白-1表達水平明顯下降,與Tseng 等[15]的研究結果一致,說明二甲雙胍可能通過下調血管p38 MAPK的表達、抑制p38 MAPK信號轉導通路,阻斷白細胞介素、腫瘤壞死因子、黏附分子及炎性介質的釋放,從而有效改善糖尿病大鼠主動脈粥樣硬化情況,發揮對糖尿病大血管病變的保護作用。
總之,p38 MAPK參與了糖尿病大血管病變的發生。通過干預p38 MAPK信號轉導通路中的某些環節可緩解糖尿病大血管病變的發生、發展。二甲雙胍除降糖作用外,還可通過下調血管p38 MAPK磷酸化水平、抑制炎癥反應、調節脂代謝,發揮對糖尿病大血管的保護作用。
[1] Liu Y, Liang C, Liu X, et al. AGEs increased migration and inflammatory responses of adventitial fibroblasts via RAGE, MAPK and NF-kappaB pathways. Atherosclerosis, 2010, 208: 34-42.
[2] Zhang R, Zhou SJ, Li CJ, et al. C-reactive protein/oxidised low-density lipoprotein/β2-glycoprotein I complex promotes atherosclerosis in diabetic BALB/c mice via p38 mitogen-activated protein kinase signal pathway. Lipids Health Dis, 2013, 12: 42-53.
[3] Lu J, Ji J, Meng H, et al. The protective effect and underlying mechanism of metformin on neointima formation in fructose-induced insulin resistant rats. Cardiovasc Diabetol, 2013, 12: 58.
[4] Ghatak SB, Dhamecha PS, Bhadada SV, et al. Investigation of the potential effects of metformin on atherothrombotic risk factors in hyperlipidemic rats. Eur J Pharmacol, 2011, 659: 213-223.
[5] Ishibashi Y, Matsui T, Takeuchi M, et al. Metformin inhibits advanced glycation end products (AGEs)-induced renal tubular cell injury by suppressing reactive oxygen species generation via reducing receptor for AGEs (RAGE) expression. Horm Metab Res, 2012, 44: 891-895.
[6] 郭嘯華, 劉志紅, 李恒, 等. 高糖高脂飲食誘導的2型糖尿病大鼠模型及其腎病特點. 中國糖尿病雜志, 2002, 10: 35-39.
[7] Kravchuk E, Grineva E, Bairamov A, et al. The effect of metformin on the myocardial tolerance to ischemia-reperfusion injury in the rat model of diabetes mellitus typeⅡ. Exp Diabetes Res, 2011, 2011: 907496.
[8] Keymel S, Heinen Y, Balzer J, et al. Characterization of macro-and microvascular function and structure in patients with type 2 diabetes mellitus. Am J Cardiovasc Dis, 2011, 1: 68-75.
[9] Zhang X, Song Y, Han X, et al. Liquiritin attenuates advanced glycation end products-induced endothelial dysfunction via RAGE/ NF-κB pathway in human umbilical vein endothelial cells. Mol Cell Biochem, 2013, 374: 191-201.
[10] 王芳, 趙學禮. 2型糖尿病大血管病變與血清脂聯素及炎癥因子關系的研究. 中國循環雜志, 2009, 24: 45-47.
[11] Yoon SJ, Yoo YW, Lee BK, et al. Potential role of HMG-CoA reductase inhibitor on oxidative stress induced by advanced glycation endproducts in vascular smooth muscle cells of diabetic vasculopathy. Exp Mol Med, 2009, 41: 802-811.
[12] Liu Z, Cao W. p38 mitogen-activated protein kinase: a critical node linking insulin resistance and cardiovascular diseases intype 2 diabetes mellitus. Endocr Metab Immune Disord Drug Targets, 2009, 9: 38-46.
[13] Stolar M. Glycemic control and complications in type 2 diabetes mellitus. Am J Med, 2010, 123: 3-7.
[14] 高巖, 紀立農, 劉曉寧, 等. 血漿中糖基化終級產物水平的升高與糖尿病周圍大血管并發癥的關系. 中國循環雜志, 2004, 19: 213-215.
[15] Tseng SC, Huang YC, Chen HJ, et al. Metformin-mediated downregulation of p38 mitogen-activated protein kinase-dependent excision repair cross-complementing 1 decreases DNA repair capacity and sensitizes human lung cancer cells to paclitaxel. Biochem Pharmacol, 2013, 85: 583-594.
Metformin Decreasing the Aortic p38 MAPK Protein Expression in Experimental Rats With Type 2 Diabetes Mellitus
CHEN Li-li, FAN Guo-qia, HAN Rui, SHI Zhi-ping, WANG Tao, ZHANG Jie, WANG Xue, ZHOU Ya-ru.
Department of Endocrinology, Third Hospital of Hebei Medical University, Shijiazhuang (050051), Hebei, China
Objective: To explore the relationship between p38 MAPK protein expression and macro vascular lesions in type 2 diabetes mellitus (DM) rats with the effect of metformin intervention.Methods: A total of 25 healthy male Wistar rats were randomly divided into 2 groups: Control group, the rats had normaldiet for 4 weeks and then received citrate buffer solution for modeling control, n=6. Experimental group, the rats received high fat diet for 4 weeks and then received streptozotocin injection for DM modeling, n=19; there were 15 rats successfully developed DM and randomly divided into 2 subgroups: DM control (DM-C) group, n=7 and DM + metformin group, n=8, in which DM rats received metformin 200 mg/kg·d for 8 weeks. At the end of intervention, intraperitoneal injections of glucose tolerance test ( IPGTT ) was performed, laboratory biochemical indexes and fasting insulin level were detected, protein expressions of nueclear factor (NF-κB), monocyte chemo-attractant protein-1(MCP-1) in the thoracic aorta were examined by immunohistochemistry.Results: ①HE staining showed that DM-C group had increased tunica intimae thickness, endothelia cell swollen, media smooth muscle disorder and collagen fiber hyperplasia.②Compared with Control group, DM-C group had increased levels of fasting insulin (20.00 ± 5.91) mIU/L vs (11.34 ± 3.88) mIU/L, NF-κB (170.22 ± 21.53) μmol/L vs (78.68 ± 12.15) μmol/L, ICAM-1 (130.59 ± 16.00) pg/ml vs (75.63 ± 19.52) pg/ml, VCAM-1 (990.19 ± 119.18) ng/ml vs (616.54 ±54.51) ng/ml, and increased TC (4.15 ± 0.41) mmol/L vs (2.18 ± 0.39) mmol/L, TG (1.83 ± 0.40) mmol/L vs (0.81 ± 0.27) mmol/L, LDL (2.53 ± 0.44) mmol/L vs (1.24 ± 0.47) mmol/L, P<0.05; while decreased AUCi (30.78 ± 11.25) mIU·L-1·h vs (47.55 ± 5.23) mIU·L-1·h, P<0.05; increased protein expressions of p38 MAPK, NF-κB and MCP-1, P<0.05.③Compared with DM-C group, DM + metformin group presented decreased levels of fasting insulin, NF-κB, ICAM-1, VCAM-1, TC, TG, P<0.05; increased aortic protein expressions of p38 MAPK, NF-κB and MCP-1; while AUCi was similar between 2 groups, P>0.05.Conclusion: The activation of p38 MAPK pathway plays an important role in DM macro angiopathy, metformin protects blood vessel via decreasing p38 MAPK level, inhibiting inflammatory reaction and regulating lipid metabolism in DM rats.
Diabetes mellitus; Atherosclerosis; p38 MAPK; Metformin
2014-09-08)
(編輯:漆利萍)
中華醫學會臨床科研專項資金(09010220177)
050051 河北省石家莊市,河北醫科大學第三醫院 內分泌二科(陳麗莉、范國洽、韓蕊、石志平、王雪、周亞茹), 心血管二科(王濤),河北省骨科生物力學重點實驗室(張潔)
陳麗莉 碩士研究生 主要從事糖尿病并發癥發病機制及干預研究 Email:chenlily163@163.com 通訊作者:周亞茹
Email:lzhouyaru@gmail.com
R541
A
1000-3614(2015)05-0487-05
10.3969/j.issn.1000-3614.2015.05.018
猜你喜歡
中老年保健(2022年5期)2022-08-24 02:35:42
中老年保健(2022年1期)2022-08-17 06:14:56
美與時代·美術學刊(2022年3期)2022-04-27 01:18:15
中老年保健(2021年5期)2021-08-24 07:07:20
中老年保健(2021年9期)2021-08-24 03:51:04
中老年保健(2021年7期)2021-08-22 07:42:16
中老年保健(2021年11期)2021-08-22 03:15:16
火花(2019年12期)2019-12-26 01:00:28
人大建設(2019年12期)2019-05-21 02:55:32
學苑創造·A版(2015年11期)2016-01-14 09:03:27
