青稞遺傳多樣性及其農(nóng)藝性狀與SSR標(biāo)記的關(guān)聯(lián)分析
2016-03-03 11:15:15孟亞雄孟祎林汪軍成司二靜張海娟任盼榮馬小樂李葆春王化俊
作物學(xué)報(bào) 2016年2期
孟亞雄 孟祎林 汪軍成 司二靜 張海娟 任盼榮馬小樂 李葆春 楊 軻 王化俊*
1甘肅省干旱生境作物學(xué)重點(diǎn)實(shí)驗(yàn)室 / 甘肅省作物遺傳改良與種質(zhì)創(chuàng)新重點(diǎn)實(shí)驗(yàn)室, 甘肅蘭州 730070;2甘肅農(nóng)業(yè)大學(xué)農(nóng)學(xué)院, 甘肅蘭州 730070;3甘肅農(nóng)業(yè)大學(xué)生命科學(xué)技術(shù)院, 甘肅蘭州 730070
?
青稞遺傳多樣性及其農(nóng)藝性狀與SSR標(biāo)記的關(guān)聯(lián)分析
孟亞雄1,2孟祎林1,2汪軍成1,2司二靜1,2張海娟1,2任盼榮1,2馬小樂1,2李葆春1,3楊軻1,2王化俊1,2*
1甘肅省干旱生境作物學(xué)重點(diǎn)實(shí)驗(yàn)室 / 甘肅省作物遺傳改良與種質(zhì)創(chuàng)新重點(diǎn)實(shí)驗(yàn)室, 甘肅蘭州 730070;2甘肅農(nóng)業(yè)大學(xué)農(nóng)學(xué)院, 甘肅蘭州 730070;3甘肅農(nóng)業(yè)大學(xué)生命科學(xué)技術(shù)院, 甘肅蘭州 730070
摘要:利用92個(gè)SSR標(biāo)記對(duì)108份青稞親本材料進(jìn)行多態(tài)性掃描, 分析其遺傳多樣性, 旨在尋找與農(nóng)藝性狀相關(guān)聯(lián)的分子標(biāo)記, 為青稞雜交組合的配制及分子標(biāo)記輔助育種提供依據(jù)。挑選48個(gè)多態(tài)性標(biāo)記進(jìn)行群體遺傳結(jié)構(gòu)分析,在此基礎(chǔ)上采用Tassel 2.1 GLM (general linear model)和MLM (mixed linear model)方法進(jìn)行標(biāo)記與農(nóng)藝性狀的關(guān)聯(lián)分析。共檢測出156個(gè)等位變異, 每個(gè)位點(diǎn)2~6個(gè)等位變異。供試群體的Shannon指數(shù)為0.6727~1.1368, 材料間遺傳相似系數(shù)為0.2250~1.0000, 平均0.7585。通過群體遺傳結(jié)構(gòu)分析將供試材料劃分成4個(gè)亞群。以GLM分析, 發(fā)現(xiàn)12個(gè)與株高、穗長、穗粒數(shù)和分蘗數(shù)相關(guān)聯(lián)的標(biāo)記, 對(duì)表型變異的解釋率分別為11.5%~17.6%、19.4%~45.4%、15.4%~22.1%和29.2%; 以MLM分析, 發(fā)現(xiàn)8個(gè)與株高、分蘗數(shù)和小穗數(shù)相關(guān)的標(biāo)記, 各標(biāo)記對(duì)表型變異的解釋率分別為31.7%~49.8%、28.1%~37.2%、22.7%~32.7%。關(guān)聯(lián)標(biāo)記分布在基因組全部6個(gè)連鎖群上。
關(guān)鍵詞:青稞; SSR; 遺傳多樣性; 群體遺傳結(jié)構(gòu); 關(guān)聯(lián)分析
本研究由國家自然科學(xué)基金項(xiàng)目(31460347), 甘肅省財(cái)政廳科研業(yè)務(wù)費(fèi)(035-041047)和國家現(xiàn)代農(nóng)業(yè)產(chǎn)業(yè)技術(shù)體系建設(shè)專項(xiàng)(CARS-05)資助。
This study was supported by the National Natural Science Foundation of China (31460347), Gansu Provincial Department of Finance Research Operating Expenses (035-041047), and the China Agriculture Research System (CARS-05).
第一作者聯(lián)系方式: E-mail: yxmeng1@163.com; Tel: 13919964113
青稞(Hordeum vulgare L. var. nudum HK. f.)是一種古老栽培作物, 主要分布在中國青藏高原、南美安地斯高原、北非高原及俄羅斯的考克薩斯等山區(qū)[1], 具有較高的營養(yǎng)價(jià)值, 是釀造工業(yè)的重要原料和發(fā)展養(yǎng)殖業(yè)的優(yōu)良飼料。在我國青藏高原地區(qū),總?cè)丝?0%以上的藏族居民都以青稞為主食, 因此青稞育種的突破性進(jìn)展對(duì)藏族地區(qū)經(jīng)濟(jì)發(fā)展和社會(huì)穩(wěn)定具有十分重要意義。和其他作物一樣, 青稞也面臨著親本遺傳基礎(chǔ)狹窄的育種困境[2], 亟需加強(qiáng)遺傳資源的引進(jìn)、創(chuàng)制及其農(nóng)藝性狀特征和遺傳基礎(chǔ)的了解與掌握。對(duì)現(xiàn)有親本材料進(jìn)行遺傳多樣性研究, 可有效減少相似遺傳背景的組合, 減輕育種的工作量, 對(duì)于親本的雜交組合配置具有重要的指導(dǎo)意義。同時(shí), 尋找與目標(biāo)性狀相關(guān)聯(lián)的分子標(biāo)記,可以為復(fù)雜數(shù)量性狀的遺傳學(xué)研究和分子標(biāo)記輔助育種奠定重要基礎(chǔ)。
分子標(biāo)記技術(shù)的快速發(fā)展為作物種質(zhì)資源的遺傳多樣性研究提供了便利。其中SSR (simple sequence repeat, SSR)標(biāo)記具有重復(fù)性好、多態(tài)性高、數(shù)量豐富、呈共顯性且廣泛分布于基因組等優(yōu)點(diǎn), 已被廣泛應(yīng)用于作物遺傳多樣性和基因發(fā)掘等研究[2-5]。通過關(guān)聯(lián)分析對(duì)供試群體候選基因進(jìn)行檢測或分子標(biāo)記掃描, 可以獲得豐富的基因位點(diǎn)及其等位基因信息, 在大麥[6]、小麥[7-8]、玉米[9-11]、水稻[12-13]、馬鈴薯[14]等作物中均有相關(guān)報(bào)道。Wu等[15]通過對(duì)188份西藏野生大麥材料的耐鹽性關(guān)聯(lián)分析, 找到2個(gè)與耐鹽基因HvCBF4相關(guān)聯(lián)的標(biāo)記。Brantestam等[16]利用22個(gè)SSR標(biāo)記對(duì)北歐和波羅的海區(qū)域197份春大麥的遺傳多樣性研究, 共檢測出191個(gè)等位變異,平均每個(gè)標(biāo)記檢測2~23個(gè)等位變異, 并且發(fā)現(xiàn), 與早期品種相比, 當(dāng)前栽培種的遺傳多樣性發(fā)生了明顯變化, 一些起源地材料的遺傳多樣性也發(fā)生了少量變異。Sun等[17]利用42個(gè)SSR標(biāo)記對(duì)中國西藏野生大麥及世界各起源地的部分大麥材料開展了遺傳多樣性研究, 同時(shí)進(jìn)行了14個(gè)數(shù)量性狀的關(guān)聯(lián)分析。賴勇等[6]用57個(gè)SSR標(biāo)記對(duì)113份大麥材料進(jìn)行關(guān)聯(lián)分析, 以GLM和MLM兩種關(guān)聯(lián)分析模型, 分別獲得9個(gè)與株高、穗長、芒長、穗粒數(shù)和小穗著生密度及6個(gè)與株高、芒長和小穗著生密度相關(guān)聯(lián)的標(biāo)記。Kraakman等[18]將AFLP和SSR標(biāo)記同148份春大麥栽培種的抽穗期、株高、葉銹病抗性、大麥黃矮病抗性進(jìn)行連鎖不平衡(linkage disequilibrium, LD)作圖,找到其相關(guān)標(biāo)記, 證明LD作圖是尋找與主效基因或QTL相關(guān)聯(lián)的合適標(biāo)記的另一種重要途徑。Ivandic 等[19-20]利用33個(gè)SSR標(biāo)記對(duì)大麥作關(guān)聯(lián)分析, 檢測出與開花時(shí)間、抗白粉病和耐水分脅迫能力等顯著相關(guān)的標(biāo)記。
關(guān)聯(lián)分析利用自然變異, 不需要花費(fèi)過多的時(shí)間和精力去構(gòu)建作圖群體, 可以廣泛地檢測遺傳變異。株高、穗粒數(shù)、穗長、芒長和小穗著生密度等農(nóng)藝性狀是傳統(tǒng)育種的主要選擇標(biāo)準(zhǔn)。利用關(guān)聯(lián)分析尋找與這些性狀顯著相關(guān)的標(biāo)記, 可以減少專門構(gòu)建群體進(jìn)行QTL定位的工作量。而當(dāng)前SSR標(biāo)記應(yīng)用于青稞農(nóng)藝性狀關(guān)聯(lián)分析研究的報(bào)道較少。本研究對(duì)108份青稞親本材料進(jìn)行遺傳多樣性分析, 確定大麥親本材料間的遺傳背景差異, 同時(shí)尋找與株高、穗長、穗粒數(shù)和分蘗數(shù)顯著相關(guān)聯(lián)的分子標(biāo)記,為后期青稞育種工作中的親本組配、等位基因發(fā)掘和標(biāo)記輔助選擇提供參考。
1 材料與方法
1.1試驗(yàn)材料及其DNA提取
以108份青稞農(nóng)家品種、野生種和栽培品種(系)構(gòu)成自然群體(表1), 由中國農(nóng)業(yè)科學(xué)院作物科學(xué)研究所、甘肅省作物遺傳改良與種質(zhì)創(chuàng)新重點(diǎn)實(shí)驗(yàn)室麥類種質(zhì)創(chuàng)新課題組提供。從每份材料取10粒飽滿種子置培養(yǎng)皿中, 在室內(nèi)避光培養(yǎng)。10 d后采黃化苗, 液氮–80℃速凍。采用CTAB法[21-22]從黃化苗嫩葉中提取各青稞材料基因組DNA, 并用紫外分光光度計(jì)法測其質(zhì)量和濃度, 然后置于–20℃冰箱保存。
1.2農(nóng)藝性狀測定方法
于2013年3月底和2014年3月底, 將所有材料種植于甘肅農(nóng)業(yè)大學(xué)黃羊鎮(zhèn)育種試驗(yàn)站親本圃,每份材料種5行, 行長為1.2 m。在成熟收獲時(shí)隨機(jī)抽樣, 每份材料取10株, 分別測定其株高、穗長、分蘗數(shù)、穗粒數(shù)、千粒重, 取平均值。
1.3SSR標(biāo)記檢測方法及遺傳多樣性分析

表1 供試大麥親本材料Table 1 Hulless barley materials used in this study

(續(xù)表1)
選取分布于大麥1H~7H染色體的92個(gè)SSR標(biāo)記進(jìn)行PCR擴(kuò)增, 擴(kuò)增程序?yàn)? 95℃ 5 min; 94℃50 s, 64~55℃ (touch-down PCR) 50 s, 72℃ 50 s, 10個(gè)循環(huán), 每個(gè)循環(huán)退火溫度降低1℃; 94℃ 50 s, 55 ℃ 50 s, 72℃ 50 s, 30個(gè)循環(huán); 72℃ 10 min, 4℃ 保存。擴(kuò)增產(chǎn)物經(jīng)8%聚丙烯酰胺凝膠電泳, 銀染顯色后拍照。擴(kuò)增結(jié)果按相同遷移率下有帶記為1, 無帶記為0; 同時(shí)記錄基因型, 以大寫字母A、B、C等表示。
應(yīng)用NTSYS-pc計(jì)算遺傳相似系數(shù)(genetic similarity, GS)并按非加權(quán)配對(duì)法(UPGMA)和SHAN程序聚類分析。用Popgene32統(tǒng)計(jì)基因頻率和Shannon指數(shù)。
1.4關(guān)聯(lián)分析
將多態(tài)性引物檢測結(jié)果以Structure 2.3.1軟件進(jìn)行群體遺傳結(jié)構(gòu)分析, 估計(jì)最佳群體組群數(shù)K, 其取值范圍為2~10, 將參數(shù)iterations設(shè)為10 000, burnin period設(shè)為100 000, 每個(gè)K值重復(fù)運(yùn)行6次, 依據(jù)似然值最大原則選取合適的K值為群體數(shù)目, 計(jì)算Q參數(shù), 將其作為協(xié)變量, 以SPAGeDi-1.3d處理基因型數(shù)據(jù)獲得個(gè)體間親緣關(guān)系Kinship矩陣[23]。采用Tassel 2.1軟件一般線性模型(general linear model, GLM)和混合線性模型(mixed linear model, MLM)兩種程序進(jìn)行關(guān)聯(lián)分析。GLM分析中, 以各親本材料的對(duì)應(yīng)Q值作為協(xié)變量, 將SSR標(biāo)記與株高、穗長、芒長、分蘗數(shù)和穗粒數(shù)等表型變異進(jìn)行回歸分析, 尋找與之相關(guān)聯(lián)的標(biāo)記, 并確定其解釋率; MLM分析中, 采用Q+K方法分析, 選擇計(jì)算每個(gè)標(biāo)記的遺傳力(calculate heritability for each marker)的方式, 分析方法選擇EM。分別運(yùn)用兩年試驗(yàn)表型數(shù)據(jù)結(jié)合分子標(biāo)記數(shù)據(jù)和群體結(jié)構(gòu)進(jìn)行標(biāo)記-性狀關(guān)聯(lián)分析, 確定關(guān)聯(lián)位點(diǎn)(P<0.01), 并計(jì)算標(biāo)記對(duì)表型變異的解釋率。
2 結(jié)果與分析
2.1SSR標(biāo)記分析
用92對(duì)SSR引物對(duì)108份青稞材料進(jìn)行擴(kuò)增, 其中有48對(duì)能夠得到清晰、重復(fù)性好的條帶, 并且擴(kuò)增結(jié)果表現(xiàn)多態(tài)性, 共檢測出156個(gè)等位變異, 變幅為2~6個(gè), 平均每個(gè)標(biāo)記3.2個(gè); 3H染色體上的8對(duì)引物檢測到的等位變異最多, 共28個(gè); 而1H上的5對(duì)引物檢測出等位變異數(shù)最少, 共15個(gè)。SSR引物的Shannon指數(shù)變幅為0.6727~1.1368, 平均1.0507 (表2)。可見, 這48對(duì)SSR標(biāo)記的檢測效率較高, 參試青稞種質(zhì)材料群體間的遺傳變異較大, 具有較為豐富的遺傳多樣性。
2.2遺傳相似系數(shù)及聚類分析
根據(jù)48個(gè)多態(tài)SSR標(biāo)記的檢測結(jié)果, 108份青稞親本材料的GS變異范圍為0.2250~1.0000。其中ZDM5163和ZDM5737以及ZDM5199和ZDM5169 的GS最大, 為1.0000, 其次是ZDM6552和ZDM5702, GS值為0.9938, GS最小的是川83-5319與甘青1號(hào), 為0.2250。按UPGMA法進(jìn)行聚類分析(圖1)表明, 在GS值為0.5106水平上聚為3個(gè)大類, 分別包含91份、13份和4份材料, 其中由肚里黃、ZDM5740、ZDM5376和ZDM6468四份材料構(gòu)成第III大類; 第II大類分為兩個(gè)亞類, 第1亞類由白青稞等9個(gè)品種構(gòu)成, 第2亞類由ZYM2263、ZDM6553、甘青1號(hào)和ZDM6171構(gòu)成。
2.3群體遺傳結(jié)構(gòu)分析
利用Structure 2.3.1軟件, 基于數(shù)學(xué)模型分析由108份材料構(gòu)成的自然群體的遺傳結(jié)構(gòu), 發(fā)現(xiàn)樣本的等位變異頻率特征類型數(shù)K呈持續(xù)增大趨勢, 當(dāng)K=4時(shí)出現(xiàn)峰值, 并呈現(xiàn)明顯的拐點(diǎn)(圖2-A)。由此將108份青稞親本分為4個(gè)類群(圖2-B), 分別包含21、41、3和43份。該自然群體結(jié)構(gòu)比較簡單, 能夠有效地降低群體結(jié)構(gòu)對(duì)關(guān)聯(lián)分析產(chǎn)生的影響。
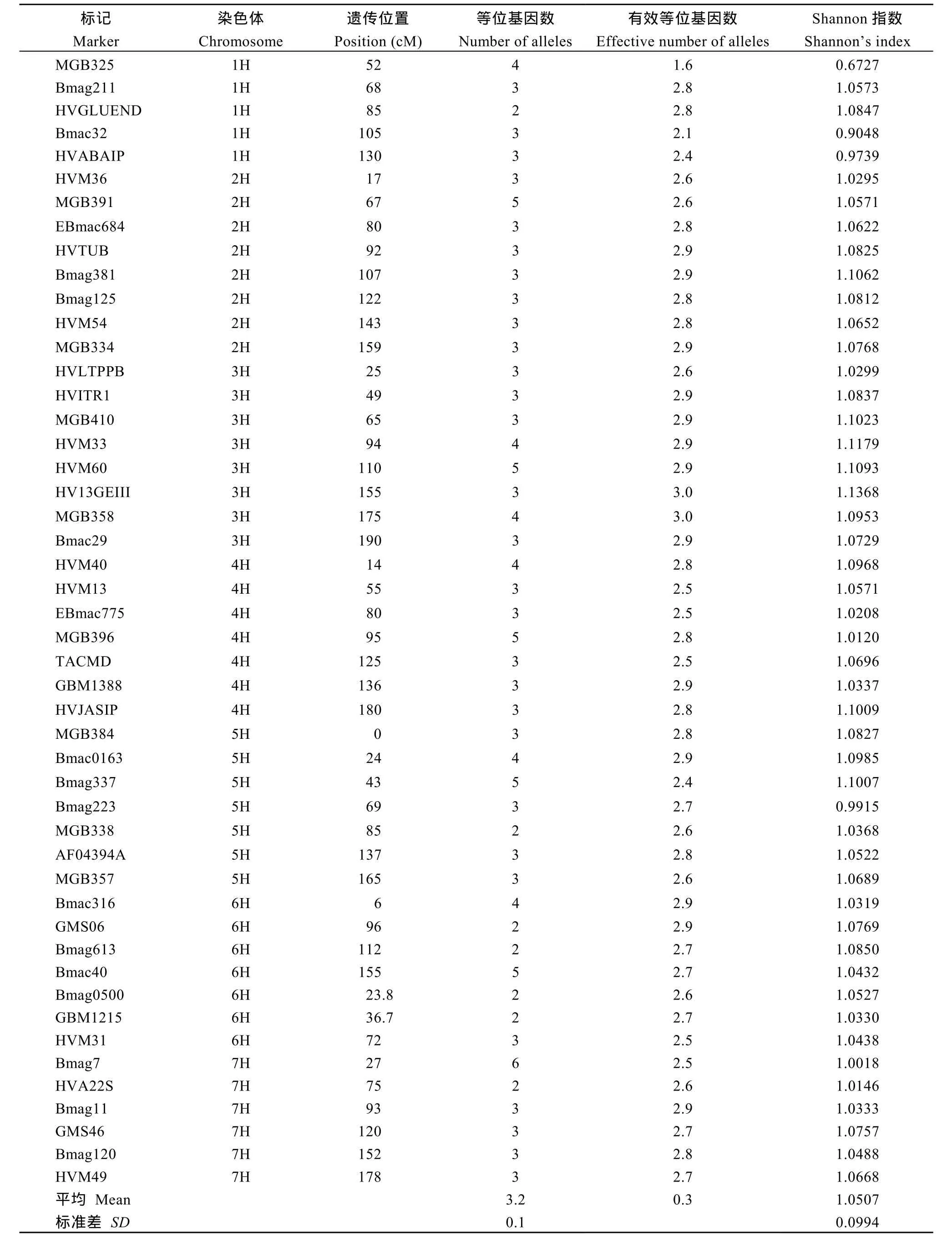
表2 48對(duì)SSR引物在108份青稞種質(zhì)材料中的多態(tài)性Table 2 Fourty-eight polymorphics SSRs among 108 accessions of hulless barley

圖1 108份青稞親本材料的聚類圖Fig. 1 Dendrogram of 108 accessions of hulless barley based on SSR markers品種編號(hào)與表1一致。Codes of varieties consistent with those given in Table 1.
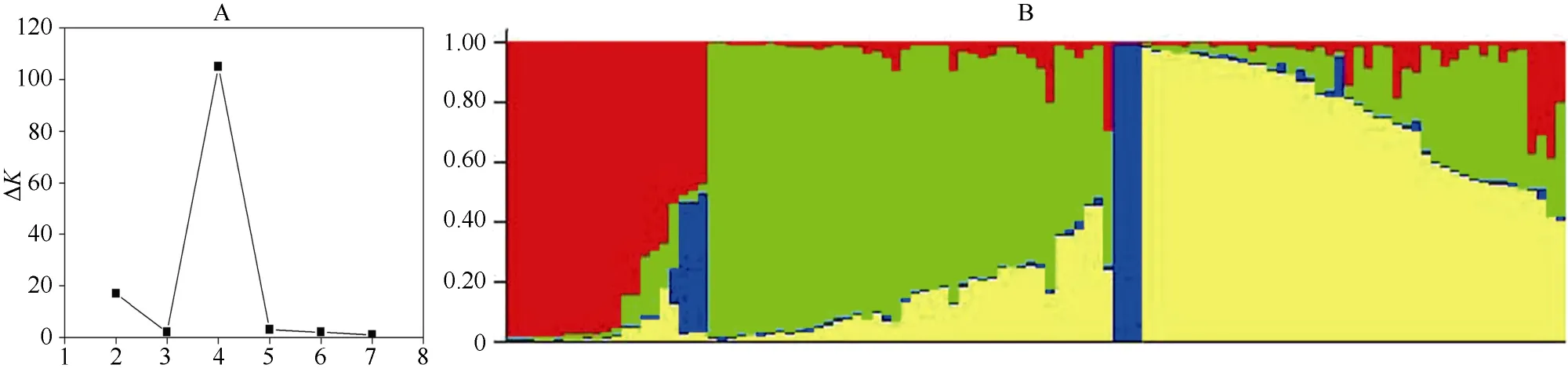
圖2 青稞親本材料的群體遺傳分析圖Fig. 2 Population genetic analysis of hulless barley accessions
2.4SSR標(biāo)記與部分農(nóng)藝性狀的關(guān)聯(lián)分析
GLM分析結(jié)果顯示, 在所檢測的48個(gè)標(biāo)記中有12個(gè)與穗長、分蘗數(shù)、穗粒數(shù)顯著相關(guān)(P<0.01)。其中6個(gè)與穗長相關(guān), 1個(gè)(EBmac775)與穗長和分蘗數(shù)都相關(guān), 3個(gè)和穗粒數(shù)相關(guān), 2個(gè)與株高相關(guān)。各標(biāo)記對(duì)表型變異的解釋率為11.5%~45.4%, 解釋率最大的標(biāo)記是HVA22S, 為45.4%, 與穗長相關(guān), 解釋率最小的標(biāo)記是Bmag381, 為11.5%, 也與穗長相關(guān)。MLM分析結(jié)果顯示, 在所檢測的48個(gè)標(biāo)記中有8個(gè)與株高、分蘗數(shù)、穗粒數(shù)顯著相關(guān)(P<0.01), 其中2個(gè)與株高相關(guān), 3個(gè)與分蘗數(shù)相關(guān), 3個(gè)與穗粒數(shù)相關(guān),各標(biāo)記對(duì)表型變異的解釋率為22.7%~49.8%, 標(biāo)記Bmac0163對(duì)株高的解釋率最大, 為49.8%, 標(biāo)記Bmac32對(duì)穗粒數(shù)的解釋率最小, 為22.7%。MLM分析檢測到與農(nóng)藝性狀相關(guān)的SSR比GLM分析結(jié)果少4個(gè), 其中有5個(gè)標(biāo)記與GLM 分析得到的相同, 但解釋率均有所下降, MLM分析未檢測到與穗長相關(guān)的標(biāo)記(表3)。
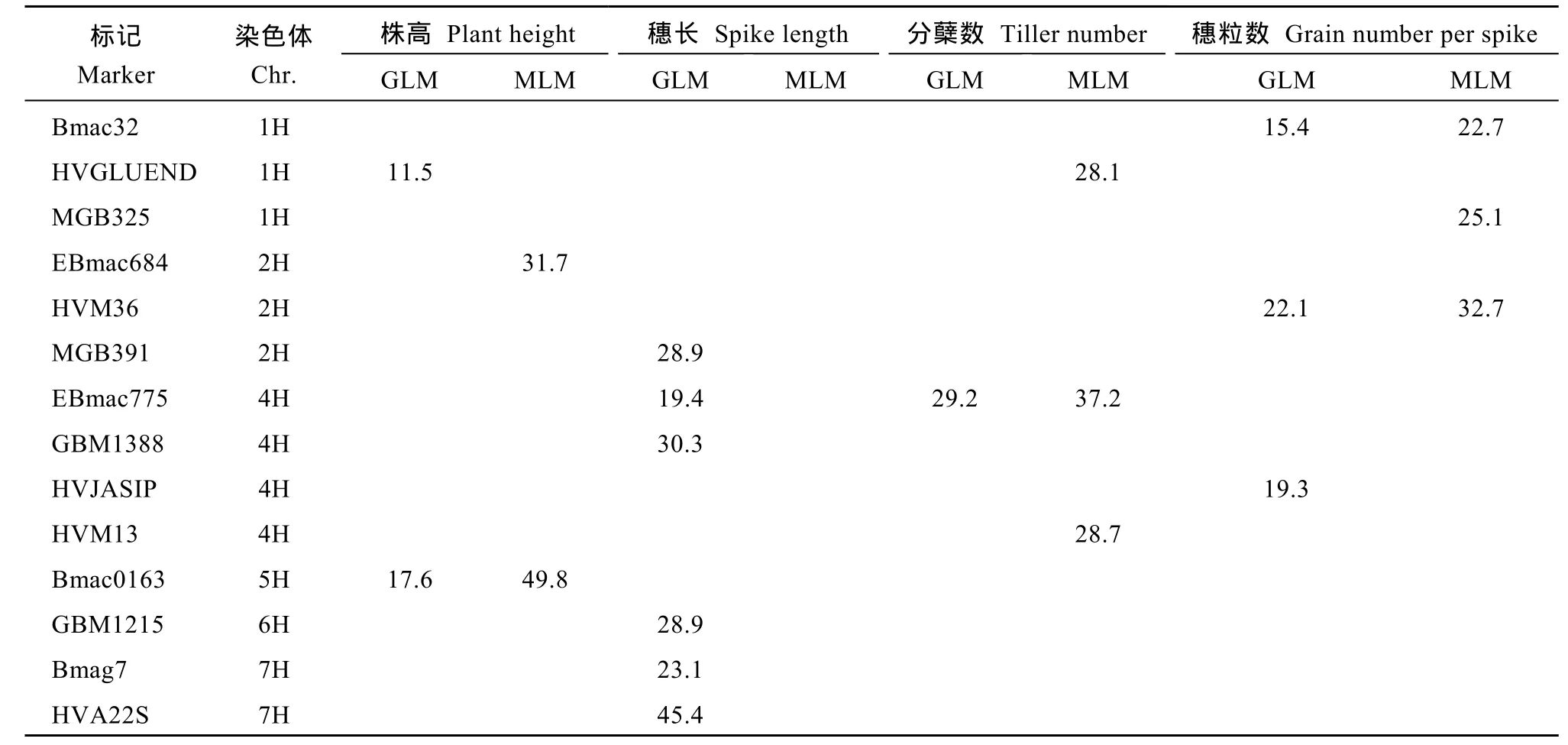
表3 與農(nóng)藝性狀相關(guān)的SSR標(biāo)記及其表型解釋率Table 3 SSR markers associated with agronomic traits and their explained phenotypic variations (%)
3 討論
3.1青稞親本材料的性狀及遺傳多樣性分析
種質(zhì)材料是進(jìn)行作物育種的基礎(chǔ), 遺傳基礎(chǔ)狹窄是制約培育出優(yōu)良品種的主要因素之一。因此分析親本材料的遺傳多樣性, 比較材料相互間親緣關(guān)系的遠(yuǎn)近, 對(duì)于開展作物育種工作具有重要的指導(dǎo)意義。本研究選用48對(duì)分布于大麥1H~7H染色體的
SSR標(biāo)記, 基本覆蓋全基因組, 能夠較全面地分析108份材料的遺傳多樣性, 比較它們遺傳背景的差異, 為親本的雜交組合合理配置在分子水平上提供依據(jù)。國內(nèi)已開展了SSR標(biāo)記檢測青稞的遺傳多樣性的研究, 如孟凡磊等[1]結(jié)合青稞育種和生產(chǎn)利用的實(shí)際, 對(duì)我國西藏主要農(nóng)區(qū)青稞品種的遺傳多樣性進(jìn)行了研究, 發(fā)現(xiàn)供試青稞材料間存在一定的遺傳差異, 但品種間的遺傳距離較近, 西藏青稞育成品種的基因來源相對(duì)較窄。潘志芬等[24]利用SSR標(biāo)記分析了64份青藏高原栽培青稞的遺傳多樣性, 認(rèn)為青藏高原栽培青稞具有豐富的遺傳多樣性。楊平等[25]利用SRAP分子標(biāo)記技術(shù), 對(duì)25份來自四川高原的青稞育成品種進(jìn)行了遺傳多樣性研究, 發(fā)現(xiàn)聚類特征與材料來源地有明顯相關(guān), 25份材料間的平均遺傳距離較小(0.3240), 平均遺傳多樣性較低(0.5126), 遺傳基礎(chǔ)較為狹窄。本研究所用的108份青稞種質(zhì)材料也存在類似的問題, 遺傳相似系數(shù)(GS)變異范圍為0.2250~1.0000, 平均為0.7585, 在GS值為0.5106水平上可將參試材料聚為3個(gè)大類群, 各大類群分別包括91、13和4份材料, 說明各材料親緣關(guān)系較近, 需要引入一些新的材料, 拓展親本遺傳基礎(chǔ), 其中品種ZDM5163和ZDM5737以及ZDM5199和ZDM5169之間的遺傳相似系數(shù)最大, GS為1.0000,表明ZDM5163和ZDM5737以及ZDM5199和ZDM5169遺傳背景可能比較相近, 本研究中出現(xiàn)兩份材料間GS值為1的情況, 原因可能是品種本身遺傳相似性較高, 或者也有可能是本研究所選取的引物數(shù)量有限導(dǎo)致, 本研究中只選用了48對(duì)引物, 引物的數(shù)量有限可能導(dǎo)致了未能完全區(qū)分供試材料間差異。川83-5319與甘青1號(hào)的GS值最小, 為0.2250,說明它們之間的遺傳背景可能比較遠(yuǎn), 可以考慮用于雜交組合的選配。
3.2青稞分子標(biāo)記的關(guān)聯(lián)分析
關(guān)聯(lián)分析通過分析作物種質(zhì)資源中標(biāo)記與緊密連鎖QTL之間的LD關(guān)系來鑒定QTL, 能夠直接對(duì)基因型變異和表型變異進(jìn)行分析, 可以確定不同種質(zhì)資源中所攜帶的等位基因及其對(duì)目標(biāo)性狀的貢獻(xiàn)[6]。對(duì)種質(zhì)資源進(jìn)行遺傳多樣性及群體遺傳結(jié)構(gòu)分析, 是關(guān)聯(lián)作圖的前提, 因?yàn)槿后w遺傳結(jié)構(gòu)會(huì)通過對(duì)連鎖不平衡位點(diǎn)LD的影響從而對(duì)關(guān)聯(lián)分析的準(zhǔn)確性產(chǎn)生影響, 而LD水平的高低是關(guān)聯(lián)分析的基礎(chǔ), 在亞群的基礎(chǔ)上對(duì)是否達(dá)到哈德溫伯格平衡的數(shù)學(xué)模型的群體結(jié)構(gòu)分析, 可以計(jì)算出相對(duì)應(yīng)的Q值(第i個(gè)材料基因組變異源于第k群體的概率), 在群體結(jié)構(gòu)中亞群的混合能使整個(gè)群體LD的強(qiáng)度增加, 從而造成偽關(guān)聯(lián)[26-29]。因此, 可以將Q值作為協(xié)變量納入回歸分析, 從而消除參試材料的群體結(jié)構(gòu)引起的偽關(guān)聯(lián),也保證了后續(xù)關(guān)聯(lián)分析的準(zhǔn)確性。所以, 對(duì)現(xiàn)有種質(zhì)資源進(jìn)行群體遺傳結(jié)構(gòu)分析能更好地體現(xiàn)單個(gè)樣本趨向各群體的比例, 對(duì)今后全面了解資源整體的遺傳信息具有重要意義。
Hansen等[30]利用AFLP標(biāo)記研究野生甜菜生長習(xí)性, 這是首次在植物中進(jìn)行全基因組關(guān)聯(lián)分析的報(bào)道。Cockram等[31]利用關(guān)聯(lián)分析對(duì)控制春化反應(yīng)的基因VRN-H1和VRN-H2分析鑒定證明, 運(yùn)用適當(dāng)?shù)慕y(tǒng)計(jì)學(xué)方法, 校正群體結(jié)構(gòu)復(fù)雜程度高的亞群劃分后, 可將該群體用于關(guān)聯(lián)分析。株高是最受重視的農(nóng)藝性狀之一, 研究表明, 在大麥1H到7H染色體上均有控制株高的QTL[6,32-34]。GrainGenes2.0網(wǎng)站(http://wheat.pw.usda.gov/GG2/index.shtml)公布的大麥株高相關(guān)標(biāo)記有88個(gè), 其中12個(gè)為SSR標(biāo)記。本研究表明108份青稞可被分成4大類群, 分別包含21、42、3和44份材料。以CLM和MLM兩種模型的關(guān)聯(lián)分析, 分別找到12個(gè)和8個(gè)與株高、穗長、分蘗數(shù)、穗粒數(shù)相關(guān)的標(biāo)記, 并且MLM中檢測到的標(biāo)記Bmac32、Bmac0163、HVGLUEND、EBmac775和HVM36在GLM中同樣被檢測到, 但標(biāo)記的解釋率較之要高。本研究與分蘗數(shù)相關(guān)的3個(gè)標(biāo)記HVGLUEND、HVM13、EBmac775分別位于1H、4H、4H上, 其中標(biāo)記EBmac775在前人研究中也被檢測到, 但與千粒重相關(guān)[35], 可能是性狀間相關(guān)的遺傳原因所致。與穗長相關(guān)的標(biāo)記GBM1215、Bmag7、MGB391、Bmag381、EBmac775、GBM1388、HVA22S分別位于6H、7H、2H、2H、4H、4H、7H上, 其中MGB391和Bmag7還曾被報(bào)道與株高相關(guān)聯(lián)[6,34]。目前與穗粒數(shù)相關(guān)標(biāo)記的文獻(xiàn)報(bào)道較少, 本研究發(fā)現(xiàn)了4個(gè)與穗粒數(shù)相關(guān)的標(biāo)記, 分別是位于1H、1H、2H 和4H上的MGB325、Bmac32、HVM36和HVJASIP, 其中HVM36與Wang等[36]報(bào)道的一致, 落在標(biāo)記區(qū)間15.8~20.9內(nèi)。
4 結(jié)論
108份材料可被分成4個(gè)大類群。并通過GLM 和MLM兩種關(guān)聯(lián)分析模型, 分別尋找到12個(gè)和8個(gè)與株高、穗長、穗粒數(shù)和分蘗數(shù)相關(guān)聯(lián)的標(biāo)記, 這些標(biāo)記位于青稞各條染色體上。
References
[1] 孟凡磊, 強(qiáng)小林, 佘奎軍, 唐亞偉, 胡銀崗. 西藏主要農(nóng)區(qū)青稞品種的遺傳多樣性分析. 作物學(xué)報(bào), 2007, 33: 1910–1914
Meng F L, Qiang X L, She K J, Tang Y W, Hu Y G. Genetic diversity analysis among hulless barley varieties from the major agricultural areas of Tibet. Acta Agron Sin, 2007, 33: 1910–1914 (in Chinese with English abstract)
[2] Tanksley S D, McCouch S R. Seed banks and molecular maps: unlocking genetic potential from the wild. Science, 1997, 277: 1063–1066
[3] Bhagwat A A, Cregan P B, Akkaya M S. Length polymorphisms of simple sequencerepeat DNA in soybean. Genetics, 1992, 132: 1131–1139
[4] Maroof M S, Biyashev R M, Yang G P, Zhang Q, Allard R W. Extraordinarily polymorphic microsatellite DNA in barley: species diversity, chromosomal locations, and population dynamics. Proc Natl Acad Sci USA, 1994, 91: 5466–5470
[5] Wang Z, Weber J L, Zhong G, Tanksley S D. Survey of plant short tandem DNA repeats. Theor Appl Genet, 1994, 88: 1–6
[6] 賴勇, 王鵬喜, 范貴強(qiáng), 司二靜, 王晉, 楊軻, 王化俊. 大麥SSR標(biāo)記遺傳多樣性及其與農(nóng)藝性狀關(guān)聯(lián)分析. 中國農(nóng)業(yè)科學(xué), 2012, 46: 233–242
Lai Y, Wang P X, Fan G Q, Si E J, Wang J, Yang K, Wang H J. Genetic diversity and association analysis using SSR markers in barley. Sci Agric Sin, 2013, 46: 233–242 (in Chinese with English abstract)
[7] Maccaferri M, Sanguineti M C, Noli E, Tuberosa R. Population structure and long-range linkage disequilibrium in a durum wheat elite collection. Mol Breed, 2005, 15: 271–290
[8] Liu S B, Yang X P, Zhang D D, Bai G H, Chao S M, Bockus W. Genome-wide association analysis identified SNPs closely linked to a gene resistant to soil-borne wheat mosaic virus. Theor Appl Genet, 2014, 127: 1039–1047
[9] Ducrocq S, Madur D, Veyrieras J B, Camus-Kulandaivelu L, Kloiber-Maitz M, Presterl T, Ouzunova M, Manicacci D, Charcosset A. Key impact of Vgt1 on flowering time adaptation in maize: evidence from association mapping and ecogeographical information. Genetics, 2008, 178: 2433–2437
[10] Kumar B, Abdel-Ghani A H, Pace J, Reyes-Matamoros J, Hochholdinger F, Lübberstedt T. Association analysis of single nucleotide polymorphisms in candidate genes with root traits in maize (Zea mays L.) seedlings. Plant Sci, 2014, 224: 9–19
[11] Wen W W, Li D, Li X, Gao Y Q, Li W Q, Li H H, Liu J, Liu H J, Chen W, Luo J. Metabolome-based genome-wide association study of maize kernel leads to novel biochemical insights. Nat Commun, 2014, 5: 34–38
[12] Eizenga G C, Agrama H A, Lee F N, Yan W, Jia Y. Identifying novel resistance genes in newly introduced blast resistant rice germplasm. Crop Sci, 2006, 46: 1870–1878
[13] Yonemaru J, Mizobuchi R, Kato H, Yamamoto T, Yamamoto E, Matasubara K, Hirabayashi H, Takeuchi Y, Tsunematsu H, Ishii T. Genomic regions involved in yield potential detected by genome-wide association analysis in Japanese high-yielding rice cultivars. BMC Genomics, 2014, 15: 346
[14] D’hoop B B, Keizer P L, Paulo M J, Visser R G, van Eeuwijk F A,van Eck H J. Identification of agronomically important QTL in tetraploid potato cultivars using a marker-trait association analysis. Theor Appl Genet, 2014, 127: 731–748
[15] Wu D Z, Qiu L L, Xu L, Ye L Z, Chen M X, Sun D F, Chen Z H, Zhang H T, Jin X L, Dai F, Zhang G P. Genetic variation of HvCBF genes and their association with salinity tolerance in Tibetan annual wild barley. PLoS One, 2011, 6: e22938
[16] Brantestam A K, Bothmer R, Dayteg C, Rashal I, Tuvesson S, Weibull J. Genetic diversity changes and relationships in spring barley (Hordeum vulgare L.) germplasm of Nordic and Baltic areas as shown by SSR markers. Genet Resour Crop Evol, 2007, 54: 749–758
[17] Sun D F, Ren W B, Sun G L, Peng J H. Molecular diversity and association mapping of quantitative traits in Tibetan wild and worldwide originated barley (Hordeum vulgare L.) germplasm. Euphytica, 2011, 178: 31–43
[18] Kraakman A W, Martnez F, Mussiraliev B, Eeuwijk F A, Niks R E. Linkage disequilibrium mapping of morphological, resistance, and other agronomically relevant traits in modern spring barley cultivars. Mol Breed, 2006, 17: 41–58
[19] Ivandic V, Hackett C A, Nevo E, Keith R, Thomas W T B, Forster B P. Analysis of simple sequence repeats (SSRs) in wild barley from the fertile crescent: associations with ecology, geography and flowering time. Plant Mol Biol, 2002, 48: 511–527
[20] Ivandic V, Thomas W T B, Nevo E, Zhang Z, Forster B P. Associations of simple sequence repeats with quantitative trait variation including biotic and abiotic stress tolerance in Hordeum spontaneum. Plant Breed, 2003, 122: 300–304
[21] Paterson A H, Brubaker C L, Wendel J F. A rapid method for extraction of cotton (Gossypium spp.) genomic DNA suitable for RFLP or PCR analysis. Plant Mol Biol Rep, 1993, 11: 122–127
[22] Porebski S, Bailey L G, Baum B R. Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol Biol Rep, 1997, 15: 8–15
[23] Earl D A. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour, 2012, 4: 359–361
[24] 潘志芬, 鄒弈星, 鄧光兵, 翟旭光, 吳芳, 余懋群. 青藏高原栽培青稞SSR標(biāo)記遺傳多樣性研究. 中山大學(xué)學(xué)報(bào)(自然科學(xué)版), 2007, 46: 82–86
Pan Z F, Zou Y X, Deng G B, Zhai X G, Wu F, Yu M Q. Genetic diversity of SSR Markers in cultivated hulless barley from Qinghai-Tibet plateau in China. Acta Sci Nat Univ Sunyatseni, 2007, 46: 82–86 (in Chinese with English abstract)
[25] 楊平, 劉仙俊, 劉新春, 李俊, 王希文, 何守樸, 馮宗云. 利用SRAP標(biāo)記研究四川高原青稞育成品種的遺傳多樣性. 遺傳, 2008, 30: 115–122
Yang P, Liu X J, Liu X C, Li J, Wang X W, He S P, Feng Z Y. Genetic diversity analysis of the developed Qingke (hulless barley) varieties from the plateau regions of Sichuan province in China revealed by SRAP markers. Hereditas (Beijing), 2008, 30: 115–122 (in Chinese with English abstract)
[26] Harris B P, Stokesbury K E. The spatial structure of local surficial sediment characteristics on Georges Bank, USA. Continental Shelf Res, 2010, 30: 1840–1853
[27] Wang M L, Zhu C S, Barkley N A, Chen Z B, Erpelding J E, Murray S C, Tuinstra M R, Tesso T, Pederson G A, Yu J M. Genetic diversity and population structure analysis of accessions in the US historic sweet sorghum collection. Theor Appl Genet, 2009, 120: 13–23
[28] Kline J B, Moore D J, Clevenger C V. Activation and association of the Tec tyrosine kinase with the human prolactin receptor: mapping of a Tec/Vav-receptor binding site. Mol Endocrinol, 2001, 15: 832–841
[29] 武玉國, 吳承來, 秦保平, 王振林, 黃瑋, 楊敏, 尹燕枰. 黃淮冬麥區(qū)175個(gè)小麥品種的遺傳多樣性及SSR標(biāo)記與株高和產(chǎn)量相關(guān)性狀的關(guān)聯(lián)分析. 作物學(xué)報(bào), 2012, 38: 1018–1028
Wu Y G, Wu C L, Qin B P, Wang Z L, Huang W, Yang M, Yin Y P. Diversity of 175 wheat varieties from Yellow and Huai River Valleys facultative wheat zone and association of SSR markers with plant height and yield related traits. Acta Agron Sin, 2012, 38: 1018–1028 (in Chinese with English abstract)
[30] Hansen M, Kraft T, Ganestam S, Sall T, Nilsson N O. Linkage disequilibrium mapping of the bolting gene in sea beet using AFLP markers. Genet Res, 2001, 77: 61–66
[31] Cockram J, White J, Leigh F J, Lea V J, Chiapparino E, Laurie D A, Mackay I J, Powell W, O'Sullivan D M. Association mapping of partitioning loci in barley. BMC Genet, 2008, 9: 16
[32] Marquez-Cedillo L A, Hayes P M, Kleinhofs A, Legge W G, Rossnagel B G, Sato K, Ullrich S E, Wesenberg D M. QTL analysis of agronomic traits in barley based on the doubled haploid progeny of two elite North American varieties representing different germplasm groups. Theor Appl Genet, 2001, 103: 625–637
[33]Teulat B, Borries C, This D. New QTLs identified for plant water status, water-soluble carbohydrate and osmotic adjustment in a barley population grown in a growth-chamber under two water regimes. Theor Appl Genet, 2001, 103: 161–170
[34] Korff M, Wang H, Léon J, Pillen K. AB-QTL analysis in spring barley: II. Detection of favourable exotic alleles for agronomic traits introgressed from wild barley (H. vulgare ssp. spontaneum). Theor Appl Genet, 2006, 112: 1221–1231
[35] 司二靜, 張宇, 汪軍成, 孟亞雄, 李葆春, 馬小樂, 尚勛武, 王化俊. 大麥農(nóng)藝性狀與SSR標(biāo)記的關(guān)聯(lián)分析. 作物學(xué)報(bào), 2015, 41: 1064–1072
Si E J, Zhang Y, Wang J C, Meng Y X, Li B C, Ma X L, Shang X W, Wang H J. Association analysis between SSR marker and agronomic traits in barley. Acta Agron Sin, 2015, 41: 1064–1072 (in Chinese with English abstract)
[36] Wang J, Yang J, McNeil D L, Zhou M. Identification and molecular mapping of a dwarfing gene in barley (Hordeum vulgare L.) and its correlation with other agronomic traits. Euphytica, 2010, 175: 331–342
URL: http://www.cnki.net/kcms/detail/11.1809.S.20151207.1041.002.html
Genetic Diversity and Association Analysis of Agronomic Characteristics with SSR Markers in Hulless Barley
MENG Ya-Xiong1,2, MENG Yi-Lin1,2, WANG Jun-Cheng1,2, SI Er-Jing1,2, ZHANG Hai-Juan1,2, REN Pan-Rong1,2, MA Xiao-Le1,2, LI Bao-Chun1,3, YANG Ke1,2, and WANG Hua-Jun1,2,*
1Gansu Provincial Key Laboratory of Aridland Crop Science / Gansu Key Laboratory of Crop Improvement & Germplasm Enhancement, Lanzhou 730070, China;2College of Agronomy, Gansu Agricultural University, Lanzhou 730070, China;3College of Life Sciences and Technology, Gansu Agricultural University, Lanzhou 730070, China
Abstract:The objectives of this study were to find molecular markers associated with yield-related traits and guide parental combination in molecular marker-assisted breeding and hybrid breeding of hulless barley (Hordeum vulgare L. var. nudum HK. f.). A natural hulless barley population composed of 108 parental varieties/lines was screened with 92 SSR markers, in which 48 markers were polymorphic. Population structure was analyzed based on the polymorphic SSR data and association between markers and five agronomic traits were performed in TASSEL GLM (general linear model) and MLM (mixed linear model) programs. A total of 156 alleles were detected in the 108 varieties/lines with 2–6 alleles per locus. The Shannon’s index of the population ranged from 0.6727 to 1.1368 and the genetic similarity between varieties ranged from 0.2250 to 1.0000, with the mean of 0.7585. Structure analysis revealed four genetic subpopulations for the entire materials tested. Based on GLM analysis, 12 SSR markers were found to be associated with plant height, spike length, grain number per spike and tiller number, with phenotypic contributions of 11.5–17.6%, 19.4–45.4%, 15.4–22.1% and 29.2%, respectively. Based on MLM analysis, 8 SSR markers were associated with plant height, awn length, and spikelet compactness, with the phenotypic contributions of 31.7–49.9%, 28.1–37.2%, and 22.7–32.7%, respectively. These associated markers were distributed on 6 chromosomes of the barley genome.
Keywords:Hulless barley; SSR; Genetic diversity; Population structure; Association analysis
收稿日期Received(): 2015-07-03; Accepted(接受日期): 2015-11-20; Published online(網(wǎng)絡(luò)出版日期): 2015-12-07.
通訊作者*(Corresponding author): 王化俊, E-mail: whuajun@yahoo.com, Tel: 13809315256
DOI:10.3724/SP.J.1006.2016.00180
