X射線熒光光譜法測定燒結礦堿度不確定度的評定
2016-03-06 08:01:42尹顯武
天津冶金 2016年6期
尹顯武
(天津天鐵冶金集團技術中心,河北涉縣056404)
X射線熒光光譜法測定燒結礦堿度不確定度的評定
尹顯武
(天津天鐵冶金集團技術中心,河北涉縣056404)
為評定X射線熒光光譜法測定燒結礦堿度的不確定度,分別對燒結礦CaO和SiO2含量的測量不確定度進行評定,再利用標準合成方式對非直接測量“堿度”的測量不確定度進行評定,指出評定過程中不確定度分量可能重復引入,給出了建議的標準物質不確定度分量。
X射線熒光光普;測量;不確定度;燒結礦;堿度
1 引言
燒結礦是高爐煉鐵最重要的原料,作為高爐配礦使用。它是將各種含鐵原料、輔料及適量的燃料和熔劑按工藝要求進行配比后,在燒結設備上進行燒結。堿度是燒結工藝的重要指標,通常使用堿性氧化物CaO與酸性氧化物SiO2的質量分數的比值表示。通過調節燒結礦中堿度,從而改變入爐原料的整體堿度,保證高爐鐵水較低的硫含量和噸鐵焦比。燒結礦堿度穩定性與燒結礦的產量和質量關系密切,因此,了解燒結礦堿度及其測定結果的不確定度對高爐冶煉順行具有十分重要的意義。
本文參照中國實驗室認可委員會編制的CNAS-GL06:2006[1]以及中國金屬協會編制的CSM01010108—2006[2]中推薦的測量不確定度評定方法,對粉末壓片—X射線熒光光譜法檢測燒結礦堿度的不確定度進行評定,旨在探討“堿度”這類合成變量的測量不確定度評定方法。
2 材料與方法
2.1 儀器和試劑
島津MXF-2400波長色散型X射線熒光光譜儀;
長春ZM型振動磨;長春YYJ型壓樣機;粉末試樣制樣環;
賽多利斯CP124S電子天平,mg。
2.2 試驗方法
使用示值誤差為0.1 mg的電子天平稱取10.0 g試樣,倒入振動磨中研磨若干分鐘,在壓樣機中加壓適當時間,制得直徑為35 mm的圓形樣片,在MXF中單次測量熒光強度。然后通過工作曲線計算燒結礦中CaO、SiO2的質量分數,最后將兩者相除,從而獲得燒結礦堿度值。
3 測量不確定度評定
評價分析測試的測量水平和測量數據的質量,一直是分析工作者和管理者關注的問題,傳統的做法是用測量的準確度和精密度來衡量,在實際應用時,不能確切地給出準確度和誤差的數值,精密度雖然能給出具體的數值,但只能表示最終測量數據的重復性,不能真正衡量其測量的可靠程度。測量不確定度是經典誤差理論發展和完善的產物,通過測量不確定度的評價來定量評價測量結果的質量,并表示測量的可信程度。測量不確定度的表示及其應用現在受到各國際組織和計量部門的高度重視[3]。
3.1 不確定度來源確認
X射線熒光光譜法測定結果可能包含不確定度來源主要有[2]:試樣稱量過程引入的不確定度;測量重復性引入的不確定度;工作曲線變動引入的不確定度;標準物質或控樣引入的不確定度;儀器校正引入的不確定度。
3.2 測量不確定度分量評定
3.2.1 稱量引入的不確定度
根據檢定證書該電子天平的示值誤差為0.1 g,采用矩形分布[1],引入的不確定度為:
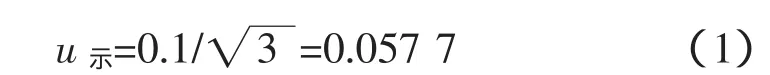
稱樣要求精確到0.1g,同樣采用矩形分布[1],引入的不確定度為:
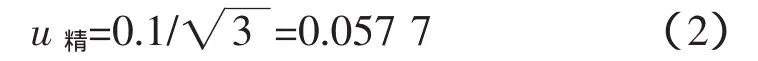
采用方和根法合成上述兩項不確定度,得到稱量引入不確定度總和[1]:

3.2.2 重復性引入的不確定度
取一袋均勻的燒結礦試樣,按試驗方法中敘述的步驟制作10枚樣片,在X射線熒光光譜儀中分析各元素含量,計算平均值、標準偏差。并按下列公式計算不確定度及相對標準不確定度。
測量不確定度[1-2]:
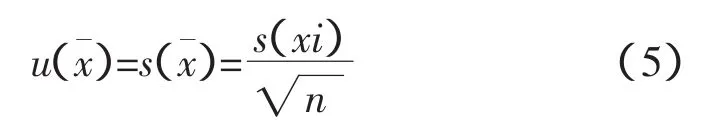
相對標準不確定度[1-2]
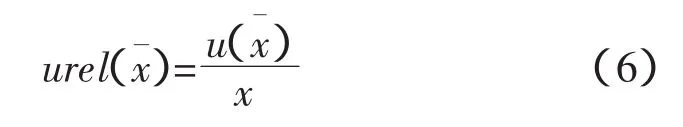
匯總結果見表1。
3.2.3 工作曲線引入的不確定度
兩種元素測定工作曲線均采用一元回歸方程:

式中,I為被測元素熒光強度,kcps;C為被測元素質量分數,%;a為回歸方程的斜率;b為回歸方程的截距。
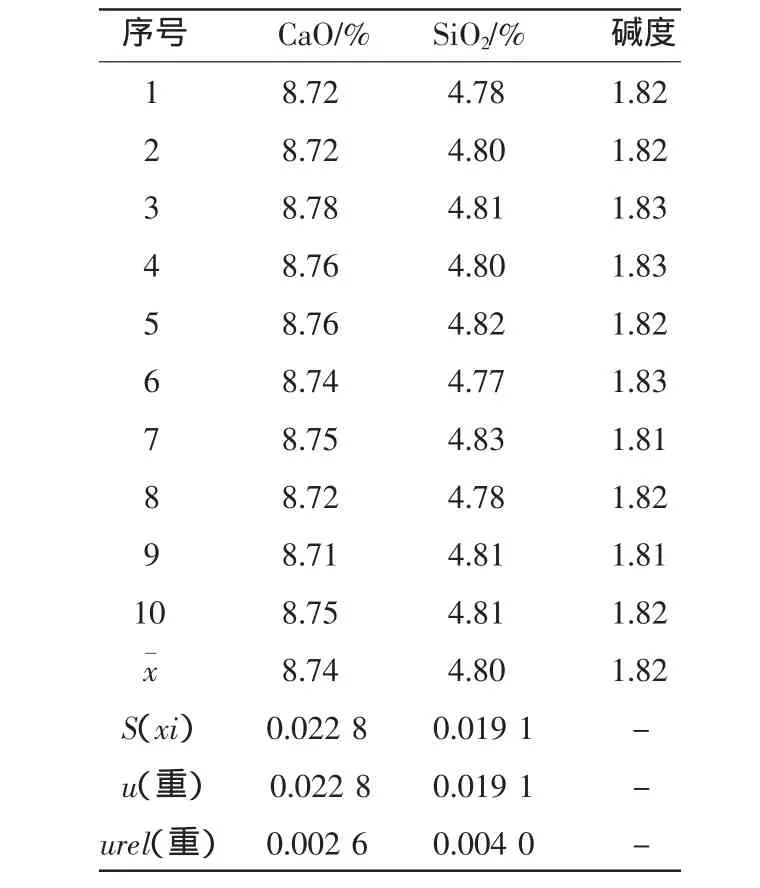
表1 重復性測量數據
CaO的工作曲線:
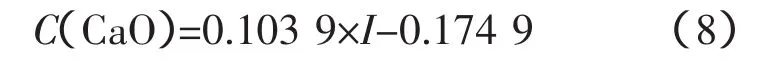
式中,a=0.103 9;b=-0.1749。
SiO2的工作曲線:
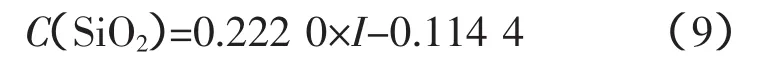
式中,a=0.222 0;b=-0.114 4。
根據參考文獻[1],工作曲線變動產生的測量不確定度為:
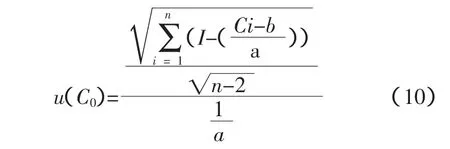
式中,P為測量C0的次數(方法采用一次測量,即P=1);n為測量標準樣品次數(n=6個×3次=18次),共6個試樣,每個樣片分別測量3次,在表2中匯總。
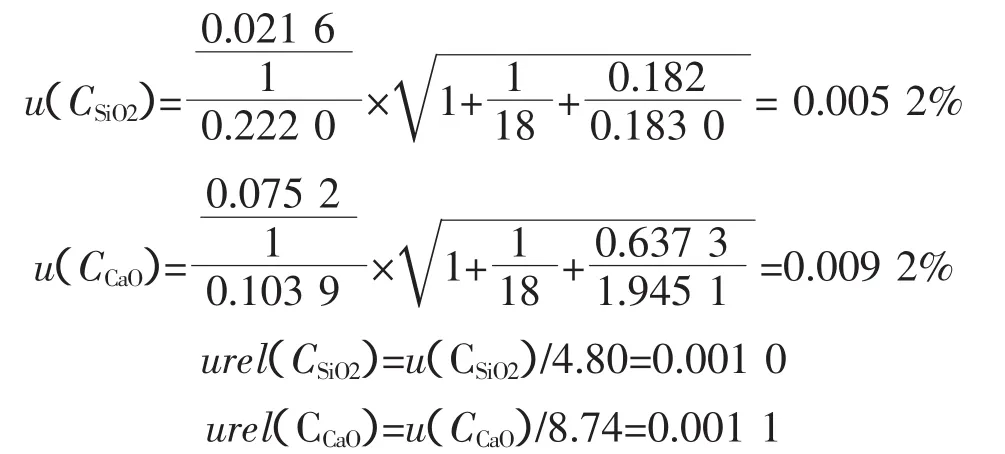
3.2.4 標準物質或控樣引入的不確定度
粉末組成均勻的壓片—X射線熒光光譜法檢測需要試樣與工作曲線試樣的集體盡可能一致,在制成片時,要求制成表面平整、光潔和良好均勻性的樣片,由于市場購買的成分和性質燒結礦便準物質不適用,我們采用化學方法多次統計平均定值,按標準控樣繪制工作曲線,樣品引入的不確定度(可用極差法)如表3、表4所示。
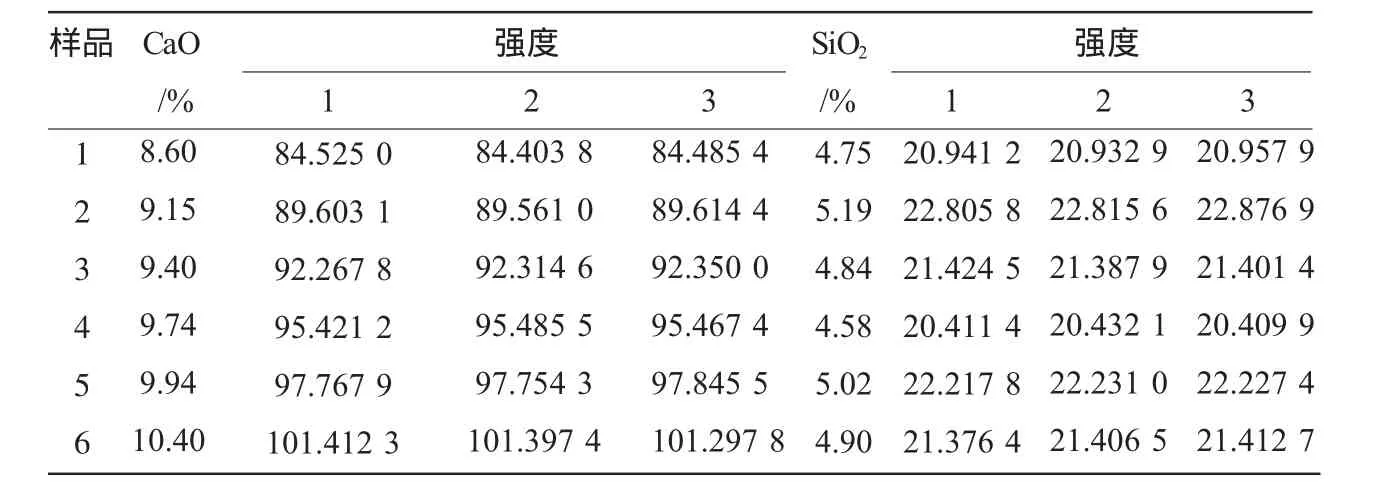
表2 樣品測量數據
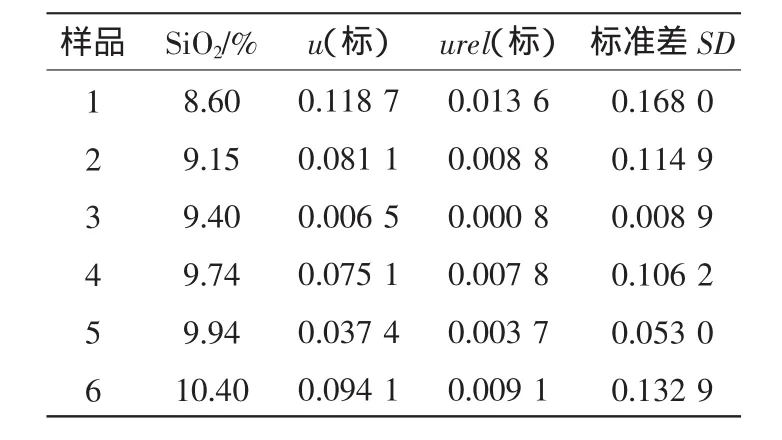
表3 標準物質中SiO2不確定度
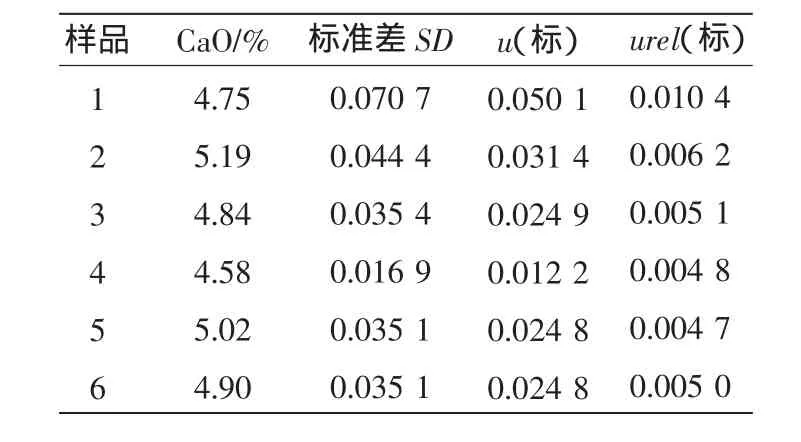
表4 標準物質中CaO不確定度
采用均方根法[2]合成上述試樣的不確定度,得到標準物質引入的不確定度總和:
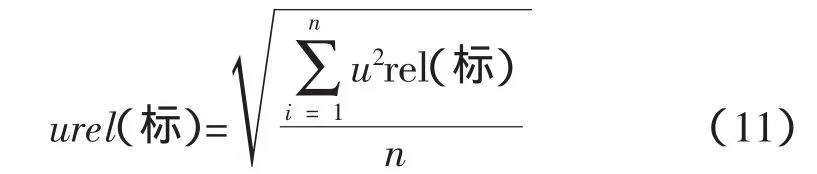
可得:
urel(標SiO2)=0.006 2
urel(標CaO)=0.008 4
3.2.5 儀器校正引入的不確定度
由于環境溫度變化較大,再加之儀器設備元件老化,都可能導致分析數據偏差,因此,每次測量前必須對儀器進行高、低標準試樣漂移校正。
為避免引入儀器內部檢測變化產生的誤差,通過對校正試樣進行3次靜態重復分析,記錄其原始強度,按3.2.2中的公式計算,結果見表5。
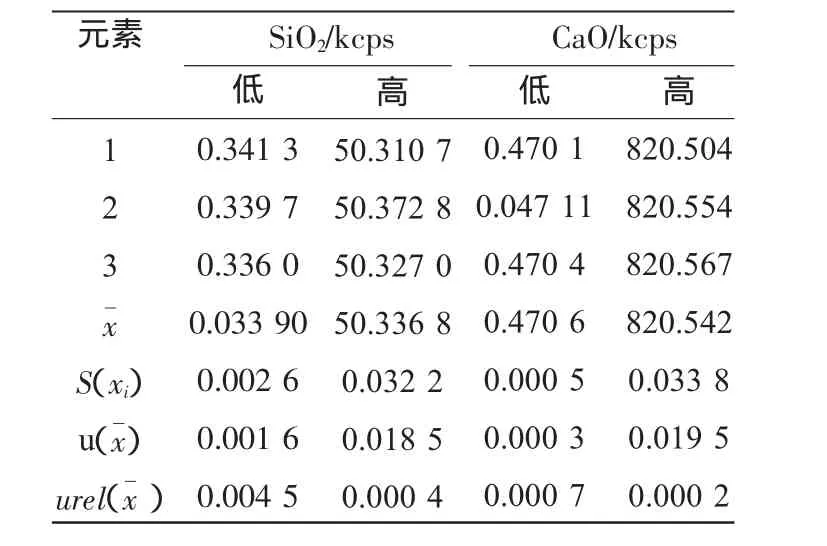
表5 靜態分析數據
校正引入的不確定度采用以下公式計算[2]:
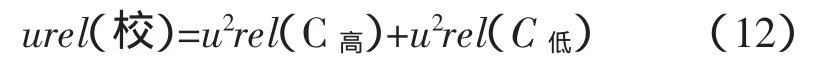
可得:CaO的校正不確定度urel(校)=0.000 5;
SiO2的校正不確定度urel(校)=0.003 3。
3.3 合成不確定度的評定
分別評定兩個元素的合成不確定度,上述討論中,各個不確定度分間沒有明顯相關性,采用方和根法合成不確定度。
3.3.1 CaO測量的合成不確定度
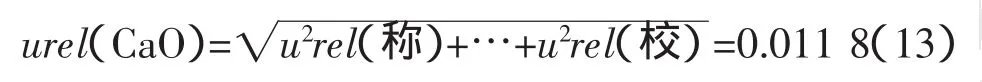
3.3.2 SiO2測量的合成不確定度
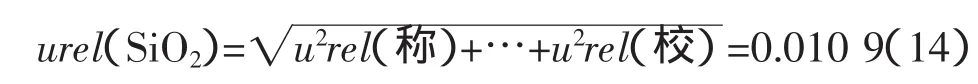
3.3.3 堿度合成不確定度的過評
在實驗過程中發現,若單一的將CaO與SiO2的合成不確定度視為堿度的不確定度分量,就會過度引入稱量誤差;稱量步驟試劑執行一次,這將導致堿度的合成不確定度過評。
由稱量引入的相對標準不確定度實際就是堿度不確定度的一個分量,不應當在3.3.1和3.3.2中引入,而應作為一個獨立分量計算。即:
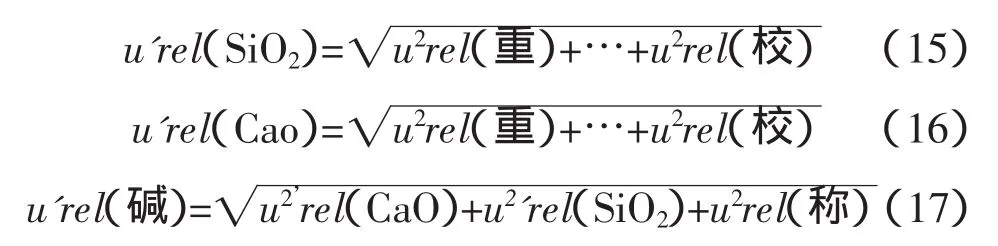
3.3.4 堿度合成不確定度的略評
采用均方根法[2]合成標準物質控樣引入的不確定度,存在一定的缺陷,繪制工作曲線實際并不能降低標準物質的不確定度,而均方根的計算方式使得不確定度被(忽)略評。建議采用標準物質或控樣中不確定度最大值作為標準物質或控樣引入的不確定度。即:
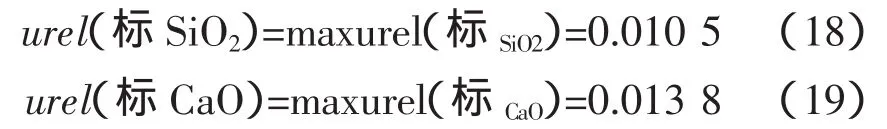
3.3.5 堿度測量結果及不確定度表示
若按文獻[2]方法進行計算,燒結礦堿度的不確定度為0.026,在95%置信區間因子K=2時,擴展不確定度為0.05;通過對標準物質引入的不確定度糾正后,燒結礦的不確定度為0.037,在95%置信區間內,包含因子K=2,則擴展不確定度為0.07.可見兩種方法評定的不確定度相差0.02。
綜上所述,粉末壓片—波長色散型X射線熒光光譜法測量燒結礦堿度的測量結果可表述為:(堿度)1.82±0.07(K=2)。
4 討論
本文的重點在于探索“堿度”這類非直接測量值的測量不確定度評定過程。建議在評價此類不確定度時,應當注意避免不確定度分量的重復引入。建議在計算標準物質或控樣引入的不確定度時,采用標準物質或控樣中最大不確定度。
[1]中國實驗室國家認可委員會.化學分析中不確定度的評估指南[M].北京:中國計量出版社,2002.
[2]CSM01010108—2006,X—射線熒光光譜法測量結果不確定度評定規范[S].
[3]曹宏燕,劉振清,蔣楊虎,等.冶金材料分板技術與應用[M].北京:冶金工業出版社,2008.
Evaluation and Analysis of Uncertainty of Sinter Ore Alkalinity Measurement with X-ray Fluorescence Spectrometry
YIN Xian-wu
(Technology Center of Tianjin Tiantie Metallurgy Group,She County,Hebei Province 056404,China)
In order to evaluate the uncertainty of sinter ore alkalinity measurement with X-ray fluorescence spectrometry,the measurement uncertainty of CaO and SiO2in sinter ore was determined and then that of" alkalinity"which was derived by non-direct measurement with standard composite method analyzed.The au-thor points out uncertainty components during evaluation process could be introduced repeatedly and proposes the uncertainty components of standard material.
X-ray fluorescence spectrometry;measurement;uncertainty;sinter ore;alkalinity
10.3969/j.issn.1006-110X.2016.06.013
2016-08-18
2016-09-03
尹顯武(1982—),男,工程師,主要從事冶金分析測試方面的研究工作。
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
中學生數理化·八年級物理人教版(2019年9期)2019-11-25 07:33:02
當代陜西(2019年8期)2019-05-09 02:22:48
中學生數理化·八年級物理人教版(2019年3期)2019-04-25 06:20:54
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
中學生數理化·八年級物理人教版(2018年3期)2018-05-31 08:52:45
家庭影院技術(2018年4期)2018-05-09 07:07:52
數學小靈通(1-2年級)(2017年10期)2017-11-08 08:39:45
少兒科學周刊·兒童版(2016年1期)2016-03-14 03:52:21
專用汽車(2016年4期)2016-03-01 04:13:43
