低碳烷烴深加工制烯烴技術的研究進展
2016-03-21 02:19:41張海娟張浩楠奚兆毅
石油化工 2016年12期
張海娟,高 杰,張浩楠,萬 海,奚兆毅
(1. 遼寧石油化工大學 化學化工與環境學部,遼寧 撫順 113001;2. 中國石油 撫順石化公司石油三廠,遼寧 撫順 113001)
技術動態
低碳烷烴深加工制烯烴技術的研究進展
張海娟1,高 杰1,張浩楠1,萬 海2,奚兆毅1
(1. 遼寧石油化工大學 化學化工與環境學部,遼寧 撫順 113001;2. 中國石油 撫順石化公司石油三廠,遼寧 撫順 113001)
低碳烷烴深加工脫氫是一種廣泛應用的大規模生產純烯烴的方式。主要綜述了低碳烷烴深加工脫氫制烯烴技術的特點及市場份額較高的兩種代表性脫氫工藝;詳細評述了國內外低碳烷烴深加工脫氫技術現狀和商業化的脫氫催化劑體系;總結了丙烷脫氫和異丁烷脫氫的區別和聯系;展望了這一技術未來的發展趨勢。未來具有經濟吸引力的低碳烷烴深加工脫氫技術是最佳的催化劑設計與反應器工程之間的良好的協同作用,而國內目前開發這一技術的首要任務是完成脫氫催化劑的國產化,率先在引進技術的裝置上實現脫氫催化劑的替代。
低碳烷烴脫氫;烯烴;脫氫工藝;脫氫催化劑
目前,北美頁巖氣的開發導致天然氣價格大幅下降[1],同時液化石油氣用作民用燃料很難為煉廠帶來更高效益,尋找液化石油氣高附加值利用的途徑、充分利用低碳烷烴(丙烷、異丁烷和正丁烷)逐漸受到人們的關注。此外,丙烯和異丁烯下游產品的不斷開發利用,也造成了全球性丙烯和異丁烯資源的匱乏。因此,采用低碳烷烴脫氫制烯烴技術拓展低碳烯烴來源、完成液化氣高附加值利用,蘊藏著良好的發展機遇和巨大的市場潛力。在20世紀七、八十年代,人們主要關注正丁烷-丁烯-丁二烯的脫氫反應,然而隨著市場及應用規模的變化,現在丙烷和異丁烷脫氫已變得越來越重要[2]。低碳烷烴脫氫可以從廉價及可利用的低成本飽和烴原料中獲得烯烴,是一種具有相當大工業化影響的方法[3]。
當前低碳烷烴深加工脫氫制烯烴的途徑主要有無氧脫氫(催化脫氫,泛指脫氫)、氧化脫氫和膜反應器脫氫[4-6]。氧化脫氫技術由于烷烴及脫氫產物易被深度氧化,距離工業化較遠;膜反應器技術也存在滲透壓降與反應床層壓降合理分配、傳質和傳熱等問題,限制其商業開發[7-8]。目前,只有國外一些公司開發的無氧脫氫技術比較成熟,已實現工業化,而國內尚無自主開發的工業化技術應用。因此,開展具有自主知識產權的無氧脫氫技術研究,盡快實現工業化,具有重大的意義。
本文綜述了低碳烷烴深加工脫氫制烯烴技術的特點及市場份額較高的兩種代表性脫氫工藝;詳細評述了國內外低碳烷烴深加工脫氫技術現狀和商業化的脫氫催化體系;總結了丙烷脫氫和異丁烷脫氫的區別和聯系。
1 低碳烷烴深加工脫氫制烯烴工藝
低碳烷烴脫氫是一種吸熱、平衡控制的反應,需要較高的溫度和較低的壓力以獲得顯著的烯烴收率[9]。平衡轉化率受熱力學限制,需要依靠提高反應溫度提高烷烴轉化率。這些特性對發展商業過程造成了一定的限制。為了達到經濟合理的單程轉化率(未反應烷烴分離成本高),反應溫度超過550 ℃;而且,烷烴碳數越少,需要的溫度越高,相關的工程問題越復雜[9]。但反應溫度的升高,有利于較輕烷烴熱裂解反應,加速焦炭的生成,造成催化劑失活,從而影響催化劑的選擇性和穩定性。為克服這些問題,研發了很多技術。例如,Lummus公司的Catofin技術就是在負壓下操作,通過降低反應器壓力提高平衡轉化率;UOP公司的Oleflex技術在初級階段也曾嘗試采用負壓操作來提高平衡轉化率,但負壓操作在工程操作方面大幅增加了能耗,進而增加了操作成本,Oleflex技術現采用微正壓操作[10-11]。
1.1 國外低碳烷烴脫氫5種工藝
自從Frey等[12-13]發現Cr基脫氫催化劑,許多企業為開發商業化脫氫工藝技術進行了大量的研究。低碳烷烴脫氫工藝在操作方面最主要的技術挑戰是:1)反應器需充分的供熱;2)控制溫度,減少裂解產物,最大化轉化率;3)催化劑的再生。不同公司有不同的解決方案,由此也誕生了不同的脫氫技術。
世界上低碳烷烴脫氫制烯烴專利技術有:UOP公司的Oleflex工藝、ABB Lummus公司的Catofin工藝、Uhde公司的Star工藝、Snamprogetti/Yarsintz公司的FBD-4工藝以及林德/巴斯夫公司的PDH工藝[9,14]。其中,市場占有率較高的是UOP公司的Oleflex工藝和ABB Lummus公司的Catofin工藝。Oleflex工藝和Catofin工藝已成為丙烷脫氫的代表工藝,而異丁烷脫氫的代表工藝則是Oleflex工藝、Catofin工藝和FBD工藝,林德/巴斯夫公司的PDH工藝只應用于一家示范工廠。
Catofin工藝采用一系列絕熱固定床反應器,脫氫和再生周期快速交替,循環操作。FBD工藝使用一個流化床反應器、一個與之連接的再生器,主要在俄羅斯使用,并且規模較小。Oleflex工藝則是由4個串聯移動床反應器和一個再生器組成,該工藝結合了兩種已成熟的工業化技術,即長鏈烷烴脫氫的Pacol工藝與Pt重整工藝中的催化劑連續再生技術。而Star和PDH工藝則是采用列管式固定床反應器,切換式再生。這些工藝和反應器設計細節見于諸多文獻報道[15-18]。
在采用的催化劑方面,各工藝也有所不同。Oleflex和Star工藝采用的是Pt貴金屬催化劑;Catofin,FBD,PDH工藝采用的是Cr-A1催化劑。但PDH工藝的第二代催化劑也已從Cr-A1催化體系改為貴金屬催化體系。
1.2 Oleflex工藝與Catofin工藝
1.2.1 Oleflex工藝
Oleflex工藝是UOP公司的烷烴脫氫專利技術,使用Pt基催化劑,利用低碳烷烴作原料,氫氣為稀釋劑,用以抑制結焦、抑制熱裂解和作載熱體維持脫氫反應溫度,微正壓操作,壓力0.2~0.5 MPa。該技術的優點是烯烴收率穩定、催化劑再生方法理想、催化劑使用壽命長、裝填量少,但移動床技術復雜,投資和動力消耗較高,對原料雜質要求較苛刻[9,14,16]。近年來,使用Pt-Sn催化劑的Oleflex工藝技術已成為脫氫技術的主導。Oleflex工藝流程見圖1。
Oleflex工藝脫氫制烯烴技術自1990年在泰國實現工業化以來,一直在持續不斷地改進。工藝方面,主要是優化設計、降低投資和減少操作費用。通過操作條件和設計的優化來提高工藝收率,重點集中于提高操作空速、減小反應器尺寸、降低待再生催化劑的焦含量。丙烷脫氫裝置規模也不斷提高,工業化初期的規模為100 kt/a左右,20世紀末期達到250 kt/a,到21世紀初期進一步提高至300~350 kt/a,從2004年開始一些400 kt/a以上的大型丙烷脫氫裝置開始建設。目前,中國公司引進的UOP公司的Oleflex工藝技術,丙烷脫氫裝置生產規模達到了600 kt/a[19]。
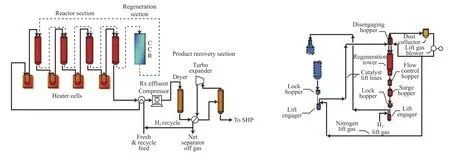
圖1 Oleflex工藝流程(反應和再生)Fig. 1 UOP Oleflex process(reaction section and regeneration section).CCR:continuous catalyst regeneration;SHP:selective hydrogenation process.
催化劑方面,則是不斷開發新一代催化劑,主要突破點為調整Pt/Sn比,降低Pt含量,提高Pt的利用率。目前己有DeH-6,DeH-8,DeH-10,DeH-12,DeH-14,DeH-16六代催化劑工業化。第一代催化劑為DeH-6Pt催化劑。1996年開發了第四代催化劑DeH-12,丙烷單程轉化率35%~40%,生成丙稀的選擇性為89%~91%。2003年位于西班牙的350 kt/a裝置已使用第五代催化劑DeH-14。目前,第六代催化劑DeH-16已用在中國投資興建的福建美德和煙臺萬華的丙烷脫氫裝置上,Pt含量降到0.3%(w)。
近三年來美國UOP公司已向中國19家生產商轉讓了Oleflex丙烷脫氫技術。自這項技術在1990年首次工業應用以來,UOP公司已交付了多套C3Oleflex和C4Oleflex工業裝置。近期,UOP公司在中國張家港新建催化劑生產基地,第一階段投資的主要產品就是丙烷脫氫工藝所用的Oleflex催化劑。
1.2.2 Catofin工藝
Catofin工藝的開發是基于20世紀40年代Houdry公司開發的丁烷生產丁二烯的Catadiene技術,采用Cr基催化劑。Catofin工藝是ABB Lummus公司開發的C3~C5烷烴脫氫生產單烯烴技術,可由丙烷脫氫制丙烯、異丁烷脫氫制異丁烯、丁烷脫氫制正丁烯和丁二烯。Catofin工藝流程見圖2[9,14,20]。
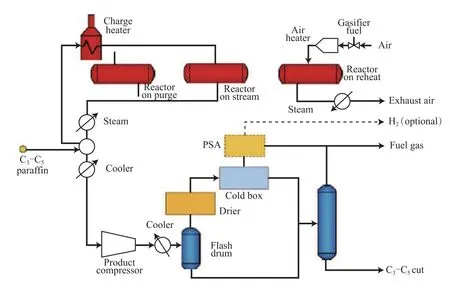
圖2 Catofin工藝流程Fig.2 Catofin process flow diagram.PSA:pressure swing adsorption.
Catofin工藝分為4個工段:脫氫制烯烴、反應器排放料的壓縮、產品的回收和精制。在一個全循環中,要進行烴蒸氣脫氫,反應器內用蒸汽清洗、空氣吹掃、預熱催化劑并燒掉少量沉積在催化劑上的結焦(基于催化劑的質量分數低于0.1),然后抽真空、復原,開始另一次循環,約23 min切換一次。為了連續生產,Catofin工藝通常采用5臺反應器,其中2臺生產、2臺催化劑再生、1臺吹掃[9,20]。
Catofin工藝技術的主要特點是:采用逆流流動循環固定床反應器,在反應器中空氣向下、烴類向上流動;使用非貴金屬催化劑,對原材料雜質要求低,價格便宜,催化劑壽命為3年,且無催化劑損失。反應器在負壓條件下操作,反應中沒有氫的再循環,沒有蒸汽稀釋,裝置運行穩定。但是,負壓操作需要不停地使用氮氣進行置換,導致這一工藝技術中氮氣的消耗非常大,而且由于催化劑單程運轉的時間非常短(約23 min),需要不停地切換。因此,Catofin工藝相對于Oleflex工藝而言,在降低能源消耗、經營成本和操作費用上并不占優勢。Catofin工藝也在不停地進行改進,反應物的入口溫度已經從最初的650 ℃降至590 ℃,可有效提高烯烴總收率。
1.3 國內低碳烷烴脫氫技術現狀
在我國,國產化的低碳烷烴深加工脫氫制烯烴技術還沒有商業應用。近二十多年來,國內有多家科研院所、高校及一些公司進行了丙烷和異丁烷脫氫的研究。中國石化的上海石化研究院、石油科學研究院和撫順石油化工研究院的研究較為深入,陸續編制了脫氫工藝包,并完成了工藝包的審查[21]。據最新報道,中國石油大學(華東)成功開發非貴金屬氧化物脫氫催化劑的丙烷/丁烷脫氫技術,在山東恒源工業試驗取得了成功。而由蘭州物理研究所開發的Al2O3負載Cr2O3型異丁烷脫氫催化劑,在東營佳昊化工進行了工業試驗。
面對低碳烷烴脫氫市場的蓬勃發展態勢,國內一些企業采用了引進技術策略。近幾年來,國內陸續引進了多套丙烷脫氫和異丁烷脫氫裝置,單丙烷脫氫總產能就超過10 Mt/a。天津渤化公司的600 kt/a丙烷脫氫、寧波海越600 kt/a丙烷脫氫、煙臺萬華600 kt/a丙烷脫氫制丙烯及衛星石化450 kt/a丙烷脫氫等裝置相繼投產,生產出合格的丙烯產品。丙烷脫氫和異丁烷脫氫大幕在中國徹底拉開,成為石化行業最耀眼的投資項目之一,多個項目預計于未來兩三年內投產。相對于丙烷脫氫而言,引進和投產的異丁烷脫氫裝置較多,并且多集中于山東省,裝置規模主要在100~300 kt/a。目前,國內已經投產的丙烷脫氫、異丁烷脫氫裝置見表1。

表1 國內已經投產低碳烷烴脫氫裝置Table 1 Domestic light paraffin dehydrogenation units which have put into production
2 低碳烷烴脫氫催化體系
低碳烷烴脫氫催化劑主要有兩類。一類是Pt基貴金屬系列催化劑,以Al2O3、鋁酸鋅或鎂鋁尖晶石做載體,Pt為主活性組分,Sn為主要助劑,如UOP開發的DH系列催化劑;另一類是Cr基氧化物系列催化劑,主要是Cr2O3/Al2O3催化劑,如Lummus公司的Cr基催化劑[22]。Cr2O3/γ -Al2O3催化劑對低碳烷烴的脫氫具有良好的活性,該催化劑對原料中雜質的要求比較低,與貴金屬催化劑相比,價格便宜,但是此類催化劑容易積碳失活。貴金屬Pt催化劑的主要優點在于活性高,選擇性較好,但是成本較高。Fe基催化劑雖然在乙苯脫氫中有很好的活性,但是在低碳烷烴脫氫反應中基本無活性[9]。商業化的低碳烷烴脫氫催化劑主要是Pt基催化劑和Cr基催化劑。
2.1 Pt基貴金屬催化劑
目前,制約Pt基催化劑廣泛應用的主要因素是其高成本和由積碳造成的催化劑失活。Pt基催化劑的研究主要集中在選擇合適的載體和助劑,以提高催化劑的性能,降低成本。國內外的公司和企業將丙烷脫氫催化劑的研究重點集中在活性組分、載體及助劑等方面的改進提高。其中,開發新型載體、添加新型助劑或對傳統的γ -Al2O3負載的貴金屬進行改進研究最為活躍[23-32]。
很多研究者嘗試使用大比表面積碳材料或介孔分子篩來實現活性組分的良好分散,提高催化劑脫氫性能。也有人嘗試從載體的酸堿性、Sn的加入方式、Pt/Sn金屬比例、Pt金屬粒徑的影響等方面進行研究,期望提高催化劑性能[32-37]。
Pt-Sn催化體系相互作用的模型如圖3所示[9]。載體的比表面積、Pt和Sn負載量、Pt/Sn比和還原溫度都影響著Pt-Sn之間的相互作用。載體比表面積減小、Pt和Sn負載量增加、Pt/Sn比降低或還原溫度過高,都會造成活性金屬與助劑狀態發生圖3中從左至右的變化,產生Pt-Sn合金,從而使催化劑失活。因此,在Pt-Sn催化體系中,催化劑載體比表面積要大、Pt和Sn負載量及Pt/Sn比例適當,還原溫度不宜過高。
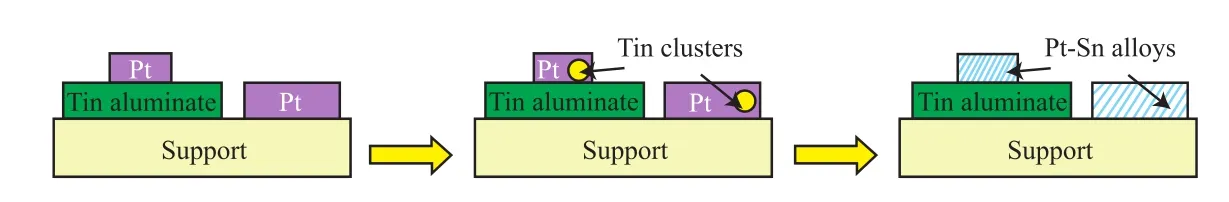
圖3 Pt-Sn相互作用模型Fig.3 Models of Pt-Sn interaction.
目前,商業化的貴金屬催化劑無法從本質上解決低碳烷烴深加工脫氫技術所遇到的瓶頸,即高烷烴單程轉化率、烯烴選擇性與催化劑積碳失活的矛盾。其根本的原因就是用于脫氫的Pt基催化劑與應用于其他領域的傳統貴金屬Pt基催化劑在Pt顆粒的分散度、粒徑分布及表面形貌等微觀結構上有著明顯的區別。如何合理設計催化劑織構、匹配活性組分與助劑是獲得高效、低積碳率的低碳烷烴Pt基催化劑的關鍵,而這方面的研究相對較少。
Pt基貴金屬脫氫催化劑失活的主要原因就是積碳。張海娟等[38]考察了低碳烷烴Pt基貴金屬催化劑的失活過程,確定了影響催化劑結焦的主要因素。對焦的芳香性質和石墨化程度來說,溫度是主要的影響因素,氫烴比次之。Afonso等[39]認為Pt基貴金屬催化劑表面積碳的形成過程主要包括以下幾個步驟:1)烴類的連續脫氫/環化;2)直鏈烴的聚合;3)Diels-Alder類反應。催化劑表面的酸量越多,越容易促進裂解、異構化等副反應的發生,增加催化劑表面的積碳量。積碳前體主要是通過金屬表面的作用(脫氫)和載體表面的作用(裂解)產生的。
李慶[40]推測了Pt基催化劑上丙烷脫氫催化劑的結焦機理,其示意圖見圖4。在丙烷脫氫制丙烯過程中,丙烷首先被吸附在催化劑的活性位上,脫氫生成炭的前體。該前體一部分生成覆蓋活性位的焦炭,另一部分可遷移至載體,參與載體上焦炭的生成。此外,生成的丙烯可直接吸附在載體的酸性位上,參與載體上的結焦過程。而在催化劑活性位上生成的焦多為不定型焦,其氫碳比相對較高;在載體上形成的焦多為石墨型焦,其氫碳比相對較低。且隨反應的進行,石墨型焦的數量大幅增長,而不定型焦的增長速度則較為緩慢。
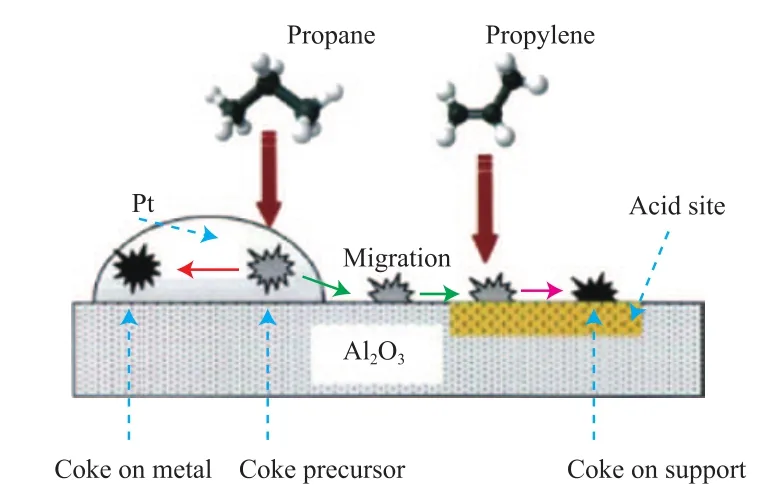
圖4 丙烷脫氫催化劑結焦機理的示意圖Fig.4 Coking mechanism on the Pt catalyst for propane dehydrogenation.
比較典型的低碳烷烴脫氫貴金屬催化劑為Oleflex工藝所采用的Pt-Sn催化劑,其發展情況見表2[16]。由表2可見,在DeH-10催化劑之后,新一代催化劑的改進主要集中于降低Pt含量、調整Sn助劑和堿金屬助劑的含量,以期活性組分與助劑之間達到最佳匹配,提高催化劑的活性和抗結焦性能。

表2 UOP 公司Oleflex工藝催化劑發展情況Table 2 Development of the dehydrogenation catalysts of the UOP Oleflex process
未來,Pt基貴金屬脫氫催化劑的設計應從活性金屬與助劑協同作用和可控的調節活性中心原子簇晶粒大小為突破點,達到提高催化劑的壽命和抗結焦性能的目的。通過調節載體表面性質,使活性組分更多地錨定于助劑界面,從而形成更多的有效活性位;對Pt顆粒進行可控制備,避免生成對C—C鍵活化效果好、有利于裂解反應的小的Pt顆粒;控制合成一定粒徑的Pt顆粒,使其對C—H鍵活化效果好,利于脫氫反應。所設計的脫氫催化劑應具有良好的目的產物選擇性、穩定性和抗結焦性能。
2.2 Cr基氧化物催化劑
由于Frey和Huppke的開創性工作,負載型Cr2O3催化劑在烷烴脫氫反應中的優越性能廣為所知,并且Cr2O3/Al2O3催化劑成為商業催化劑的廣泛選擇[12]。Frey和Huppke最初的Cr基脫氫催化劑是通過鋁和Cr的硝酸鹽和氫氧化銨的共沉淀法制備的,現在更多改良的制備方法已經開發出來[41]。
早期,Poole等[42]詳細地總結了Cr2O3/γ-Al2O3催化劑的主要性能。隨后,許多學者研究了CrOx/ SiO2,CrOx/Al2O3,CrOx/ZrO2催化劑[43-46]。研究主要集中在:1)活性位的識別;2)對Cr2O3形態的制備和處理條件以及催化劑活性、選擇性的影響;3)脫氫機理研究。
Cr系氧化物催化劑的活性和穩定性受活性位點與環境的相互作用的影響很大。活性物種的氧化狀態已成為許多年來討論和爭議的話題。很多學者認為Cr系氧化物催化劑脫氫的活性中心是配位不飽和的Cr3+,關于何種類型的Cr3+是脫氫活性中心沒有一致的結論。以Puurunen等[47]為代表的學者認為非還原Cr3+和Cr3+簇的催化活性比還原Cr3+和獨立的Cr3+要高。而以Derossi等[45]和Hakuli等[48]為代表的學者提出單核還原Cr3+是活性中心。但是也有部分學者提出Cr2+為活性物種。Gorriz等[49]研究認為,Cr3+、Cr5+和Cr6+形成多種化合物,并具有不同的還原性和催化行為。Cr5+物種和催化劑的初活性相關,但主活性中心是Cr2+,而丙烯的選擇性主要由Cr3+物種決定。
目前,越來越多的學者開始大量研究Cr系催化劑上烷烴脫氫機理。但是由于反應條件、催化劑測試手段不同,關于烷烴脫氫反應的活化、決速步驟等方面存在著很大的差異[48]。目前比較一致的看法是,烷烴脫氫反應在Cr催化劑上主要由3個步驟來完成,如圖5所示[44]。由圖5可知,首先烷烴在不飽和的Crn+中心吸附,Crn+可以是孤立的或聚集的(Crn+簇團);其次C—H鍵斷裂,O—H鍵和Cr—C鍵形成,最后在催化劑表面形成丙烯;最后丙烯從活性中心表面釋放,并且生成氫氣,催化劑活性位復原。低碳烷烴脫氫的脫氫機理研究,是低碳烷烴轉化利用理論研究的基礎,對低碳烷烴轉化利用具有重要的指導意義。
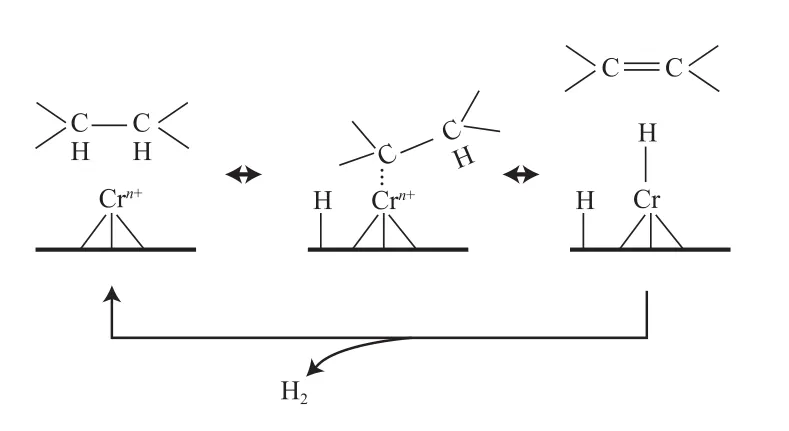
圖5 Cr2O3/Al2O3催化劑烷烴脫氫的反應機理Fig.5 Mechanism for the alkane dehydrogenation over the Cr2O3/Al2O3catalyst.
商業化的Cr2O3/Al2O3催化劑面臨最主要的問題就是催化劑失活。Cr2O3/Al2O3催化劑的失活可能由以下原因造成[48]:1)Cr活性中心燒結;2)活性中心被積碳覆蓋,阻礙了脫氫反應的進行;3)與Al2O3結合形成不具有催化活性的Cr3+物種,這是催化劑永久失活的主要原因。圖6為Cr2O3-Al2O3作用失活模型[9]。由圖6可以看出,焙燒使孤立的Cr3+離子形成Cr3+簇團,Al2O3載體的燒結誘捕Cr3+離子進入載體中,與Al2O3的Al3+空位結合,形成一種新的穩定的α-Cr2O3-Al2O3型尖晶石結構,從而喪失了催化活性[22]。這是由于Cr3+和Al3+具有相似的離子半徑和電荷數,促進了α-Cr2O3-Al2O3型尖晶石結構的形成。
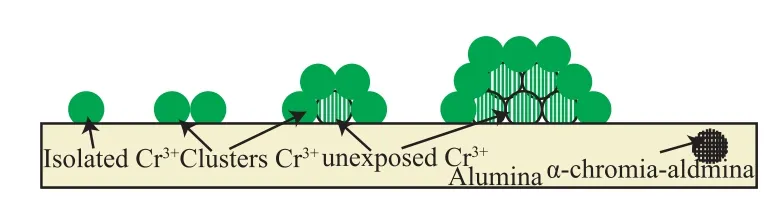
圖6 Cr2O3-Al2O3作用模型Fig.6 Model of the chromia-alumina interaction.
最成功的商業化的Cr系氧化物催化劑就是Catofin工藝所使用的Cr2O3/Al2O3催化劑,由Süd Chemie提供。催化劑以Al2O3為載體,負載Cr2O3,具有良好的機械強度,較強的抗中毒能力,可以耐受烯烴含氧化合物和重金屬。Catofin工藝技術商業化催化劑如圖7所示。

圖7 Catofin工藝應用的Cr2O3/Al2O3催化劑Fig.7 Chromia-alumina catalyst for the Catofin process.
由于Cr系催化劑對原料中雜質的要求比較低,反應活性高且價格低廉,原料易得,與貴金屬催化劑相比具有成本優勢。但是,Cr系催化劑中Cr是重金屬組分,環保治理費用較高,且穩定性差,在反應中催化劑失活較快需要反復頻繁再生,工業操作相對麻煩。因此,未來開發抗結焦且低負載量的Cr系催化劑是目前低碳烷烴脫氫技術面臨的巨大挑戰和亟待解決的問題。
3 丙烷脫氫與異丁烷脫氫技術的比較
烷烴脫氫(CnH2n+2CnH2n+H2)是強吸熱反應,單程轉化率受熱力學平衡限制[50-51]。熱力學表明,對于烷烴,碳鏈越長,脫氫越容易,所需反應溫度越低。因此,相比丙烷脫氫,異丁烷脫氫所需溫度較低。但是,丙烷和異丁烷脫氫的催化體系和脫氫工藝是類似的。由于脫氫溫度較低,異丁烷脫氫催化劑單程運轉周期較長,積碳量小。
C4烴類具有較多同分異構體,因此異丁烷脫氫產物比較復雜,而丙烷脫氫烯烴產物比較單一,目的異丁烯的選擇性要高于目的產物丙烯的選擇性。異丁烷脫氫產物流直接進入甲基叔丁基醚單元,省卻了異丁烯分離提純的過程,而丙烷脫氫產物流則要進行深冷分離,最低分離溫度接近-100 ℃,分離費用昂貴。因此從投資的角度看,相同規模的異丁烷脫氫裝置的投資要相對低一些。
國外的異丁烷脫氫技術應用多集中在20世紀80年代末期、90年代初期,2000年后期投產的異丁烷脫氫裝置鮮有報道,但是隨著國內對低碳烷烴高附加值利用的關注和重視,中國投資興建的異丁烷脫氫裝置達十余套。在國外已建的異丁烷脫氫工業裝置中,與丙烷脫氫采用的技術相反,Catofin工藝市場占有率略高于Oleflex工藝,而前蘇聯則多采用FBD工藝,這與時代大環境和那個時期的技術特點有關。在我國的低碳烷烴脫氫市場中,引進的丙烷和異丁烷脫氫技術采用Oleflex工藝較多。
綜合比較經濟性、能耗及安全環保等因素,Oleflex技術在丙烷脫氫應用市場中占有一定優勢。但是對異丁烷脫氫而言,Oleflex技術、Catofin技術及FBD技術分庭抗禮。受低油價的影響,在目前市場中,由于丙烯短缺,丙烷脫氫裝置開工率好于異丁烷脫氫裝置。
4 低碳烷烴脫氫技術未來發展趨勢
目前,低碳烷烴脫氫技術開發所面臨的問題主要有兩個方面:從催化劑角度講,降低貴金屬和Cr2O3的負載量,同時提高催化劑的活性、穩定性和抗結焦性,延長催化劑的再生周期和獲得穩定的再生性能;從工藝工程方面,就是如何實現超低壓力的低碳烷烴脫氫工藝,同時降低后期的分離能耗。
傳統的催化劑很難在高溫、低壓、高空速條件下實現催化劑的高穩定性。國際上現采用添加稀釋氣氫氣和負壓操作來達到提高催化劑活性和穩定性的目的,這些技術沒有從本質上解決催化劑脫氫活性和抗結焦性之間的矛盾。從現有烷烴脫氫技術研究趨勢看,高烷烴脫氫活性、低催化劑積碳速率是目前的催化劑研究熱點,也是從根本上解決催化劑脫氫活性與抗結焦性之間矛盾的有效方法之一。而未來具有經濟吸引力的低碳烷烴深加工脫氫技術應是最佳的催化劑設計與反應工程之間良好的協同。國內開發這一技術的首要任務是完成低碳烷烴脫氫催化劑的國產化,率先在引進技術的裝置上實現脫氫催化劑的替代,為盡快實現這一技術國產化邁出堅實的一步。
[1] Tallman M J,Klavers R. North American olefin producers riding the shale gas wave[J]. Hydrocarbon Process,2013,4:37-40.
[2] Carra S,Forni L. Catalytic dehydrogenation of C4hydrocarbons over chromia-alumina[J]. Cat Rev-Sci Eng,1971,5(1):159-198.
[3] Weckhuysen B M,Wachs I E,Schoonheydt R A. Surface chemistry and spectroscopy of chromium in inorganic oxides[J]. Chem Rev,1996,96(8):3327-3350.
[4] Chang Jongsan,Roh Hyunseog,Park Seok Min,et al. Propane dehydrogenation over a hydrogen permselective membrane reactor[J]. Bull Korean Chem Soc,2002,23(5):674-678.
[5] Bhasin M M,McCain J H,Vora B V,et al. Dehydrogenation and oxide hydrogenation of paraf fi ns to ole fi ns[J].Appl Catal,A,2001,221(1/2):397-419.
[6] 張海娟,李江紅,張喜文,等. Pt-Sn催化劑上異丁烷催化脫氫反應宏觀動力學模型[J].石油化工,2010,39(11) :1228-1231.
[7] Quicker P,Hollein V,Dittmeyer R. Catalytic dehydrogenation of hydrocarbons in palladium composite membrane reactors[J].Catal Today,2000,56(1):21-34.
[8] Miachon S,Dalmon J A. Catalysts in membrane reactors:What about the catalyst?[J].Top Catal,2004,29(1/2):59-65.
[9] Sanfilippo D,Miracca I. Dehydrogenation of paraffins:Synergies between catalyst design and reactor engineering[J].Catal Today,2006,111(1/2):133-139.
[10] 肖錦堂. 烷烴催化脫氫生產C3~C4烯烴工藝(之一) [J].天然氣工業,1994,14(2):64-69.
[11] 肖錦堂. 烷烴催化脫氫生產C3~C4烯烴工藝(之二)[J].天然氣工業,1994,14(3):69-73.
[12] Frey F E,Huppke W F. Equilibrium dehydrogenation of ethane,propane,and the butanes[J]. Ind Eng Chem,1933,25(1) :54-59.
[13] Phillips Petroleum Co. Processes for converting hydrocarbons:US2098959[P].1937-11-16.
[14] Vora B V. Development of dehydrogenation catalysts and processes[J].Top Catal,2012,55(19/20):1297-1308.
[15] 嚴樂平. 丙烷脫氫制丙烯生產技術的應用前景[J].上海化工,2010,35(7):22-27.
[16] 張琦,隋志軍,顧雄毅,等. 丙烷脫氫分離工藝的模擬與分析[J].石油化工,2015,44(4):421-428.
[17] 周巍. 丙烷脫氫制丙烯技術淺析 [J].石油化工設計,2013,30(3):36-38.
[18] Nawaz Z,Chu Y,Yang W,et al. Study of propane dehydrogenation to propylene in an integrated fluidized bed reactor using Pt-Sn/Al-SAPO-34 novel catalyst[J]. Ind Eng Chem Res,2010,49(10):4614-4619.
[19] 劉淑鶴. 丙烷脫氫反應熱力學分析和動力學研究[D].撫順:遼寧石油化工大學,2009.
[20] 朱義才. 丙烷脫氫制丙烯技術經濟分析[J].當代石油石化,2012,212(8):36-42.
[21] 王培超,曹世凌,伍寶州. 丙烷脫氫制丙烯技術的工業應用探討[J].中外能源,2015,30(5):85-89.
[22] 劉喬,董秀芹,余英哲,等. 丙烷無氧脫氫制丙烯工藝和催化劑的研究進展[J].石油化工,2014,43(6):713-720.
[23] Razavian M,Fatemi S. Synthesis and application of ZSM-5/ SAPO-34 and SAPO-34/ZSM-5 composite systems for propylene yield enhancement in propane dehydrogenation process[J]. Microporous Mesoporous Mater,2015,201:176-189.
[24] Shohreh M K,Razavian F M. Hierarchical SAPO-34 catalytic support for superior selectivity toward propylene in propane dehydrogenation process[J]. Korean J Chem Eng,2015,32(7):1289-1296.
[25] Ren Yingjie,Wang Jie,Hua Weiming,et al. Ga2O3/HZSM-48 for dehydrogenation of propane:Effect of acidity and poregeometry of support[J]. J Ind Eng Chem,2012,18(2):731-736.
[26] Long Liuliu,Lang Wanzhong,Yan Xi,et al. Yttrium-modified alumina as support for trimetallic PtSnIn catalysts with improved catalytic performance in propane dehydrogenation[J]. Fuel Process Technol,2016,146:48-55.
[27] Zhang Yiwei,Zhou Yuming,Huang Li,et al. Structure and catalytic properties of the Zn-modified ZSM-5 supported platinum catalyst for propane dehydrogenation[J].Chem Eng J,2015, 270:352-361.
[28] Biloen P,Dautzenberg F M,Sachtler W M H. Catalytic dehydrogenation of propane to propylene over platinum and platinum-gold alloys[J]. J Catal,1977,50(1):77-86.
[29] Kogan S B,Schramm H,Herskowitz M. Dehydrogenation of propane on modified Pt/θ-alumina:Performance in hydrogen and steam environment[J]. Appl Catal,A,2002,208(1/2):185-191.
[30] Zhou S,Zhou Y,Shi J,et al. Synthesis of Ce-doped mesoporous γ-alumina with enhanced catalytic performance for propane dehydrogenation[J]. J Mater Sci,2015,50(11):3984-3993.
[31] Liu Lei,Deng Qingfang,Liu Yuping,et al. HNO3-activated mesoporous carbon catalyst for direct dehydrogenation of propane to propylene[J].Catal Commun,2011,16(1):81-85.
[32] Kumar S M,Holmen A,Chen De. The influence of pore geometry of Pt containing ZSM-5,Beta and SBA-15 catalysts on dehydrogenation of propane[J].Microporous Mesoporous Mater,2009,126(1/2):152-158.
[33] Zangeneh T F,Taeba A,Gholivandc K,et al. The effect of mixed HCl-KCl competitive adsorbate on Pt adsorptionand catalytic properties of Pt-Sn/Al2O3catalysts in propane dehydrogenation[J]. Appl Surf Sci,2015,357(Part A):172-178.
[34] Liu Jie,Liu Changcheng,Ma Aizeng,et al. Effects of Al2O3phase and Cl component on dehydrogenation of propane[J]. Appl Surf Sci,2016,368:233-240.
[35] Zhang Yiwei,Zhou Yuming,Qiu Anding,et al. Propane dehydrogenation on PtSn/ZSM-5 catalyst:Effect of tin as a promoter[J].Catal Commun,2006,7(11):860-866.
[36] Kumar M S,Chen D,Holmen A,et al. Dehydrogenation of propane over Pt-SBA-15 and Pt-Sn-SBA-15:Effect of Sn on the dispersion of Pt and catalytic behavior[J].Catal Today,2009,142(1/2):17-23.
[37] Caeiro G,Carvalho R H,Wang X,et al. Activation of C2-C4alkanes over acid and bifunctional zeolite catalysts[J]. J Mol Catal A:Chem,2006,255(1):131-158.
[38] 張海娟,王振宇,李江紅,等. 反應條件對丙烷脫氫催化劑積炭行為的影響[J],天然氣化工,2014,39(2):38-42.
[39] Afonso J C,Aranda D A G,Schmal M,et al. The chemistry of coke deposits formed on a Pt-Sn catalyst during dehydrogenation of n-alkanes to mono-olefins[J]. Fuel Process Technol,1994,41(1):13-25.
[40] 李慶. Pt催化劑上丙烷脫氫反應與結焦動力學[D].上海:華東理工大學,2012.
[41] 譚曉林,馬波,張喜文,等. Cr系丙烷脫氫催化劑研究進展[J].化工進展,2010,29(1):51-57.
[42] Poole C P,Maclver D S. The physical-chemical properties of chromia-alumina catalysts[J]. Adv Catal,1967,17:223-314.
[43] Hakuli A. Preparation and characterization of supported CrOxcatalysts for butane dehydrogenation[D]. Finland:Helsinki University of Technology,1999.
[44] Weckhuysen B M,Schoonheydt R A. Alkane dehydrogenation over supported chromium oxide catalysts[J]. Catal Today,1999,51(2):223-232.
[45] Derossi S,Ferraris G,Fremiotti S,et al. Propane dehydrogenation on chromia/silica and chromia/alumina catalysts[J]. J Catal,1994,148(1):36-46.
[46] Cutrufello M G,De Rossi S,Ferino I,et al. Characterisation and activity of chromia-zirconia catalysts for propane dehydrogenation[J].Thermochim Acta,2005,434(1/2):62-68.
[47] Puurunen R L,Weckhuysen B M. Spectroscopic study on the irreversible deactivation of chromia/alumina dehydrogenatio catalysts[J].J Catal,2002,210(2):418-430.
[48] Hakuli A,Kyt?kivi A,Krause A O I,et al. Initial activity of reduced chromia/alumina catalyst in n-butane dehydrogenation monitored by on-Line FT-IR gas analysis[J].J Catal,1996,161(1):393-400.
[49] Gorriz O F,Cadu L E. Supported chromium oxide catalysts using metal carboxylate complexes:Dehydrogenation of propane[J]. Appl Catal,A,1999,180:247-260.
[50] 張海娟,李江紅,王振宇,等. 丙烷脫氫反應的熱力學分析[J].石油化工,2012,41(增刊):86-88.
[51] 張海娟,張舒冬,李江紅,等. 異丁烷脫氫反應的熱力學分析[J].化學通報,2011,74(7):628-632.
(編輯 王 馨)
Progresses in processes and catalysts for dehydrogenation of light paraffins to olefins
Zhang Haijuan1,Gao Jie1,Zhang Haonan1,Wan Hai2,Xi Zhaoyi1
(1.Chemical Engineering and Environmental Engineering,Liaoning Shihua University,Fushun Liaoning 113001,China;
2. Fushun Petrochemical Company Refinery NO.3,CNPC,Fushun Liaoning 113001,China)
The dehydrogenation of light paraffins was widely applied in the large-scale production of olefins. The characteristics of the dehydrogenation technologies were reviewed. Among them,the Oleflex process and Catofin process were illustrated due to their high market share. Two catalyst systems for the dehydrogenation processes were discussed. The difference between the propane dehydrogenation and the iso butane dehydrogenation were summarized. It was predicted that,the favourable synergy between the catalyst design and the reactor engineering would be the development trend of the light paraffin dehydrogenation in future. The most urgent task was the investigation and production of domestic dehydrogenation catalysts.
dehydrogenation of light paraffins;olefins;dehydrogenation process;dehydrogenation catalysts
1000-8144(2016)12-1411-09
TQ 203.2
A
10.3969/j.issn.1000-8144.2016.12.001
2016-08-24;[修改稿日期]2016-09-11。
張海娟(1973—),女,遼寧省葫蘆島市人,博士,副教授,電話 024-56865303,電郵 zhj_w@163.com。
猜你喜歡
山東冶金(2019年6期)2020-01-06 07:45:54
世界農藥(2019年2期)2019-07-13 05:55:12
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
銅業工程(2015年4期)2015-12-29 02:48:39
新疆鋼鐵(2015年3期)2015-11-08 01:59:52
應用化工(2014年3期)2014-08-16 13:23:50
