分階段加料熔融法制備Fe1-xO基氨合成催化劑的研究
2016-03-25 03:42:15黃仕良程田紅韓文鋒劉化章
肥料與健康 2016年6期
關(guān)鍵詞:催化劑
黃仕良,程田紅,韓文鋒,劉化章
(浙江工業(yè)大學(xué)工業(yè)催化研究所 浙江杭州 310032)
分階段加料熔融法制備Fe1-xO基氨合成催化劑的研究
黃仕良,程田紅,韓文鋒,劉化章
(浙江工業(yè)大學(xué)工業(yè)催化研究所 浙江杭州 310032)
采用熔融法制備Fe1-xO基氨合成催化劑,根據(jù)氧化物助催化劑的熔點(diǎn)、晶型和酸堿性等性質(zhì),考察了催化劑制備過程中助催化劑按順序分階段加料的方式對(duì)催化劑活性及熱穩(wěn)定性的影響。結(jié)果表明,助催化劑的添加順序?qū)Υ呋瘎┌焙铣苫钚杂幸欢ǖ挠绊懀恢呋瘎┌雌渌釅A性順序加入對(duì)催化劑氨合成活性有一定的影響,其中先加堿性助催化劑制備的催化劑的耐熱穩(wěn)定性高于其他方法制備的催化劑;助催化劑釩和鎢以其鈣鹽形式引入比以鉀鹽形式引入所得催化劑的耐熱活性高。
熔融法 氨合成 Fe1-xO基催化劑 制備方法
氨合成熔鐵催化劑的制備曾提出過多種方法,如拜爾法[1]、燒結(jié)法[2- 3]、沉淀法[4- 6]、浸漬法[7- 9]等,但工業(yè)催化劑的制備依然采用了熔融法[10]。
熔鐵催化劑是各種催化劑中制備方法最簡單的一種,其制備過程實(shí)際上就是“鍋煮”式,即將所有的原料全部混合均勻后一次性加入電熔爐[11- 12],除了生成的氣體揮發(fā)逸出外,所有的固體物料都將留在最終產(chǎn)品中,只要確保加入的生產(chǎn)原料的數(shù)量和質(zhì)量,尤其是原料的純度,并用零價(jià)鐵粉調(diào)節(jié)Fe2+/Fe3+即可滿足要求。此過程看似非常簡單,但卻包含著眾多的化學(xué)反應(yīng)、熔融與凝固、結(jié)晶與相變、晶粒生長和溶質(zhì)再分布、偏析等物理化學(xué)過程。特別是Fe1-xO基催化劑的發(fā)現(xiàn),由于其在制備原理、化學(xué)組成、晶體結(jié)構(gòu)、化學(xué)物理性質(zhì)等方面與傳統(tǒng)的Fe3O4催化劑完全不同,因此提出了新的科學(xué)問題。Fe1-xO基熔鐵催化劑是我國獨(dú)創(chuàng)的、擁有自主知識(shí)產(chǎn)權(quán)的創(chuàng)新成果,應(yīng)該有一個(gè)新的理論予以支撐。
在理論研究中發(fā)現(xiàn)[13- 15]:①熔鐵催化劑表面存在酸堿協(xié)同效應(yīng);②根據(jù)助催化劑與Fe1-xO化學(xué)物理結(jié)構(gòu)匹配原則能實(shí)現(xiàn)結(jié)構(gòu)摻雜,摻雜合適的氧化物并結(jié)合高溫快速凝固技術(shù)可有效抑制Fe1-xO的岐化反應(yīng);③H2的強(qiáng)化學(xué)吸附現(xiàn)象與催化劑表面酸堿覆蓋度有關(guān);④CaO是主要結(jié)構(gòu)助催化劑,而Al2O3雖然不是主要結(jié)構(gòu)助催化劑,但起著表面重構(gòu)作用。
關(guān)于熔融法制備氨合成催化劑的制備工藝,如熔融溫度、熔融時(shí)間以及熔漿冷卻速率等制備條件對(duì)催化劑性能的影響,國內(nèi)外學(xué)者做了大量研究。試驗(yàn)研究表明[10],隨著熔融時(shí)間的延長,F(xiàn)e2+/Fe3+呈現(xiàn)先增大后減小的趨勢(shì),故需嚴(yán)格控制熔融時(shí)間來制備合適Fe2+/Fe3+的催化劑。Lendzion- Bieluń等[16- 17]在研究熔融法制備催化劑過程中,發(fā)現(xiàn)助催化劑在催化劑中的分布與熔漿冷卻速率有很大關(guān)系,冷卻速率越慢,催化劑中Al2O3的分布越不均勻,得到的催化劑熱穩(wěn)定性差。本文提出了通過助催化劑按堿性到酸性順序分階段加料的方式來改進(jìn)“鍋煮”式熔融法制備氨合成催化劑,以期通過考察助催化劑氧化物的熔點(diǎn)、晶型和酸堿性來研究其分階段加入次序?qū)Υ呋瘎┌焙铣苫钚缘挠绊懀⒖疾熘呋瘎┭趸锴膀?qū)體對(duì)催化劑性能的影響。
1 試驗(yàn)部分
1.1 催化劑制備
根據(jù)氧化物助催化劑的酸堿性、熔點(diǎn)以及晶型等物化性質(zhì)的不同,將物料分批加入電熔爐,一部分助催化劑與精選磁鐵礦粉、還原鐵粉混合均勻后先加入電熔爐中熔融,待達(dá)到熔融狀態(tài)后再將另一部分助催化劑加入。電熔爐電源由50 kV·A鹽浴爐變壓器供給,熔融最大電流達(dá)2 700 A,最高熔融溫度為1 600 ℃。熔融一段時(shí)間后,將高溫熔漿迅速傾注入帶水夾套的冷卻槽中冷卻至室溫,再經(jīng)破碎、篩分至所需粒度。
1.2 催化劑活性評(píng)價(jià)
催化劑活性評(píng)價(jià)在內(nèi)徑為Φ14 mm的連續(xù)流動(dòng)的高壓固定床反應(yīng)器中進(jìn)行,催化劑粒度為1.0~1.4 mm,并將催化劑裝填在反應(yīng)器等溫區(qū)內(nèi)。氫氮混合氣(體積比3∶1)由液氨分解制得,經(jīng)凈化除去未分解的NH3,H2O,CO及CO2等雜質(zhì)氣體,凈化后的氣體采用壓縮機(jī)升壓。催化劑在氫氮混合氣壓力為5 MPa、空速30 000 h-1和溫度400~500 ℃條件下還原28 h,然后在15 MPa、空速30 000 h-1條件下測(cè)定反應(yīng)器出口氣體中氨體積分?jǐn)?shù),再升溫至500 ℃恒溫16 h,然后在相同條件下重新測(cè)定催化劑活性,并考察催化劑的耐熱穩(wěn)定性。
1.3 催化劑表征
NH3- TPD表征:樣品量0.5 g,粒度為150~250 μm(100~60目),樣品在流量30 mL/min、溫度110 ℃的Ar氣氛下處理1 h,除去其中的水分;然后降溫至50 ℃,以流量30 mL/min通入含氨體積分?jǐn)?shù)為10%的氨氦混合氣,恒溫0.5 h;切換Ar吹掃1 h;以10 ℃/min升溫至800 ℃。
XRD表征:催化劑還原前后的物相用瑞士Thermo ARL X′TRA型X射線衍射儀測(cè)定,Cu Kα光源(λ=1.540 56×10-10m),電壓45 kV,電流40 mA,掃描速度0.20°/min。
2 試驗(yàn)結(jié)果與分析
2.1 按氧化物的熔點(diǎn)和晶型次序添加對(duì)催化劑活性的影響
磁鐵礦的熔融溫度在1 550~1 600 ℃,隨著FeO的生成,爐溫將逐漸降低,直至穩(wěn)定在FeO的熔點(diǎn)1 400 ℃左右。助催化劑Al2O3,CaO,K2O等的熔點(diǎn)雖然高于1 600 ℃,但當(dāng)熔融物處于液體狀態(tài)時(shí),其均以離子狀態(tài)存在,理論上應(yīng)該是均勻分布的,而熔融溫度、熔融時(shí)間等因素只能影響熔融物處于液體狀態(tài)時(shí)助催化劑的分布狀態(tài)。一旦熔漿離開電熔爐而發(fā)生凝固,則助催化劑在催化劑中的分布狀態(tài)就基本上被確定,即在凝固過程中會(huì)發(fā)生溶質(zhì)再分布而產(chǎn)生偏析,造成分布不均勻。為了使這些助催化劑在催化劑中分散得更均勻,改變助催化劑的添加次序可以減少偏析,如:助催化劑與磁鐵礦粉、還原鐵粉一起加入爐中,則可延長助催化劑在電熔爐中的擴(kuò)散時(shí)間;而助催化劑V2O5,WO3,MoO3等在高溫下都存在顯著的升華現(xiàn)象,故在催化劑制備的第2階段加入,盡量縮短其在電熔爐中的時(shí)間,以減少因升華而造成的損失。
Al2O3中的Al3+能夠與Fe3+發(fā)生陽離子取代[18- 22]而形成取代型固溶體,同時(shí)也能與FeO形成化合物型固溶體;Mg2+不僅能夠取代Fe2+進(jìn)入Fe1-xO晶格中,而且MgO可在0%~100%范圍內(nèi)與FeO形成完全固溶體,能均勻地分布在催化劑母體中;CaO可與FeO形成替換固溶體[21]。因此,Al2O3,CaO及MgO都能與FeO形成固溶體,能在催化劑中很好地分散,添加次序不會(huì)影響其在催化劑中的分布情況,但考慮到其均是高熔點(diǎn)氧化物,故先投料會(huì)更有利,這也與筆者前期的試驗(yàn)結(jié)果一致。MnO,CoO及FeO同為立方晶系,只有在高溫下容易形成固溶體,為了使其在催化劑中均勻分布,應(yīng)在制備催化劑的第2階段加入,即在完全生成FeO的狀態(tài)下添加MnO或CoO。
因?yàn)楦鶕?jù)助催化劑氧化物的熔點(diǎn)和晶型確定助催化劑的加入順序,所以本試驗(yàn)主要考察WO3分別與CoO和MnO作為二次添加和一次性加入法制備的催化劑的氨合成活性進(jìn)行了比較,催化劑耐熱前后活性隨溫度變化如圖1所示,各催化劑的XRD示意如圖2所示。
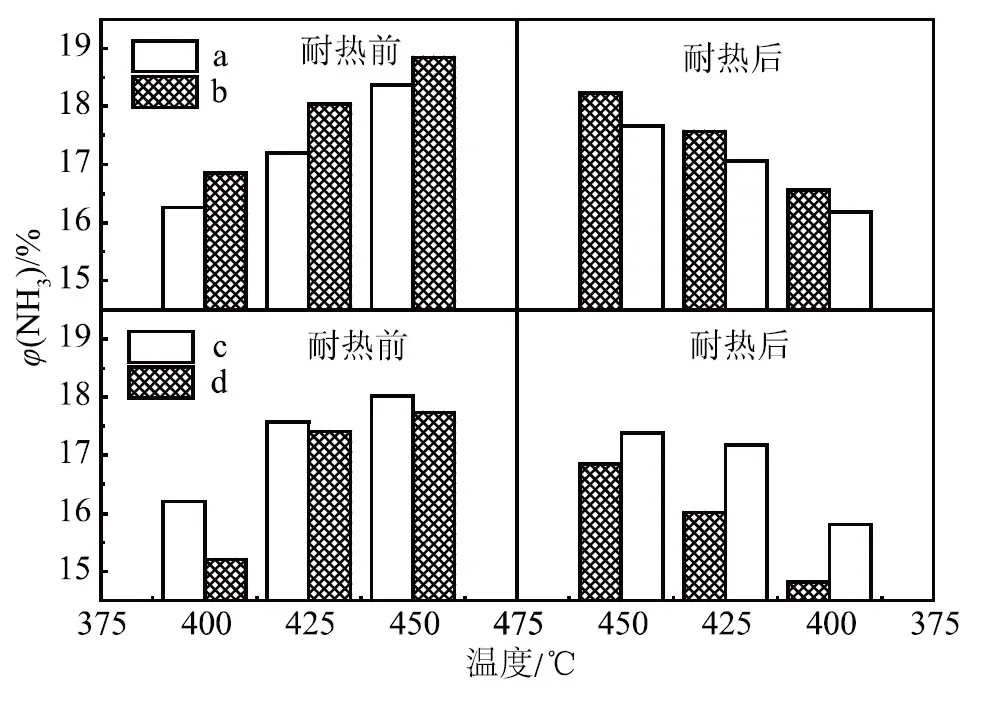
a. 一次性添加 b. 分次加入(WO3和CoO后加)c. 一次性添加 d. 分次加入(WO3和MnO后加)圖1 催化劑耐熱前后活性隨溫度變化
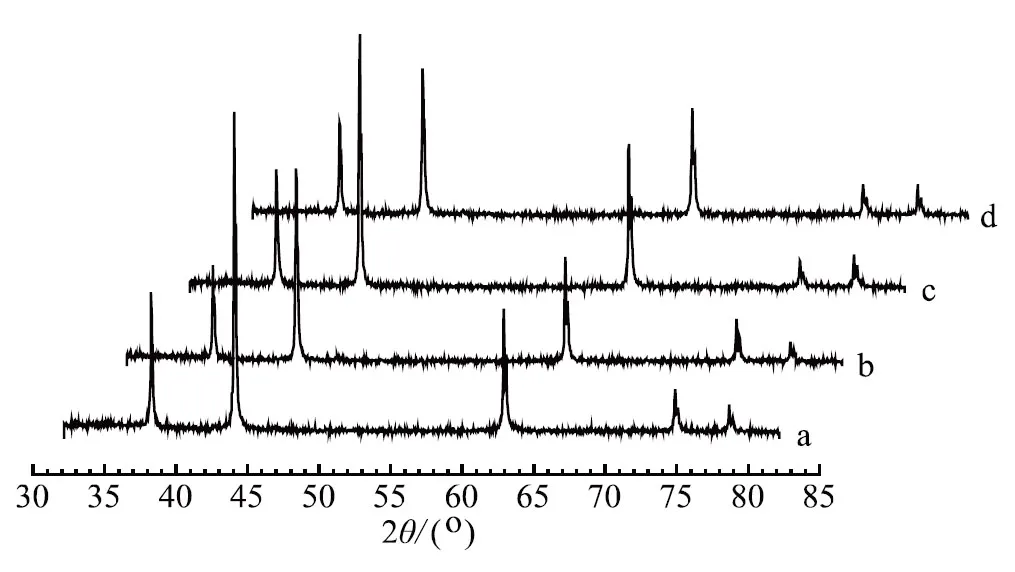
a. 一次性添加 b. 分次加入(WO3和CoO后加)c. 一次性添加 d. 分次加入(WO3和MnO后加)圖2 各催化劑的XRD示意
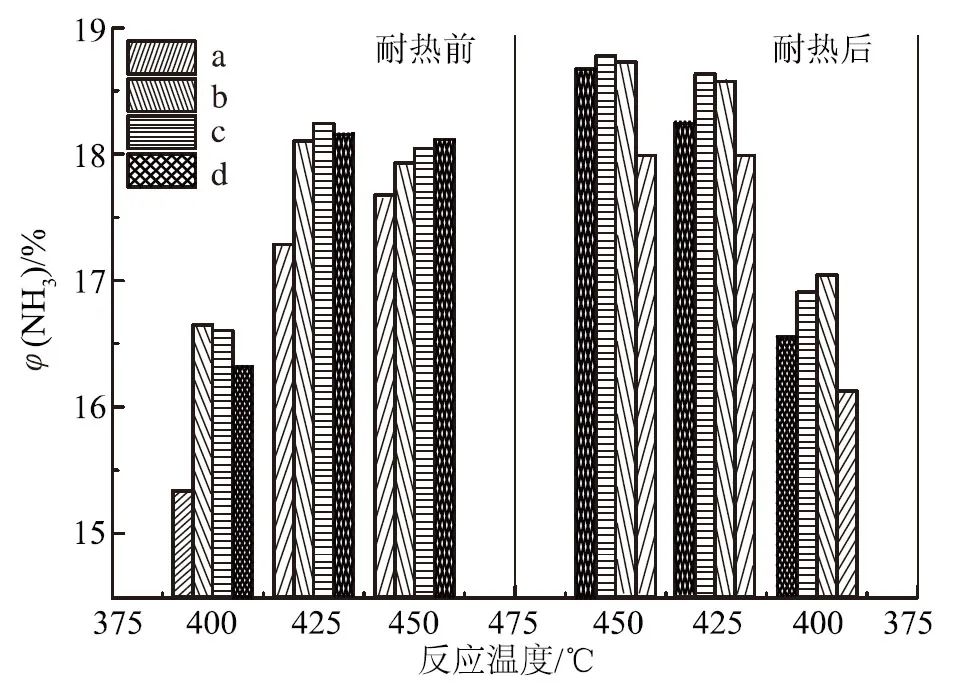
由圖1可知:助催化劑WO3和CoO后加所得催化劑耐熱前后的活性都高于一次性添加所得催化劑的活性;而助催化劑WO3和MnO的試驗(yàn)結(jié)果完全相反,即WO3和MnO一次性添加所得催化劑耐熱前后的活性都高于分次加入所得催化劑的活性,在450,425和400 ℃這3個(gè)溫度點(diǎn)的氨合成活性均是如此。由此可見,添加助催化劑的順序?qū)Π焙铣纱呋瘎┑幕钚跃哂幸欢ㄓ绊憽?/p>
由圖2可知,4個(gè)樣品的XRD圖譜中只有FeO相,并未檢測(cè)到其他物相,其主要原因是添加的助催化劑量少且在催化劑中分散均勻。采用謝樂公式計(jì)算FeO不同晶面的晶粒度如表1所示,4個(gè)樣品FeO(111)晶粒度基本一致,但FeO的另外2個(gè)主峰晶粒度差異很大,這意味著在制備催化劑過程中FeO的各晶面生長各向異性,是導(dǎo)致催化劑的氨合成活性不同的可能原因之一。
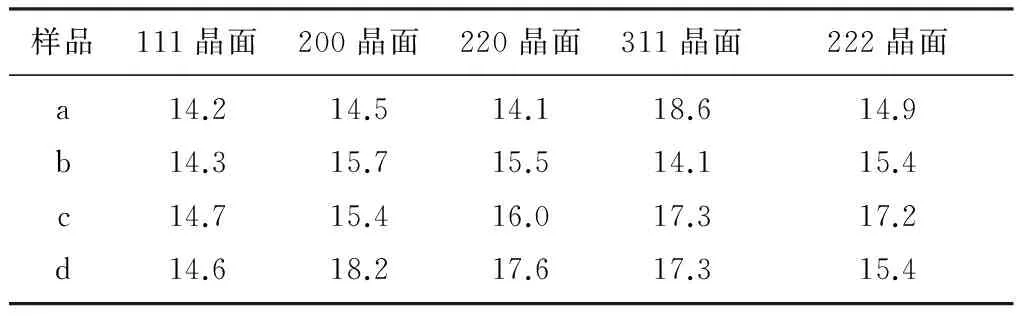
表1 FeO不同晶面的晶粒度 nm
2.2 氧化物的酸堿性對(duì)加入次序及催化劑活性的影響
劉化章等[14]在Fe1-xO基催化劑的研究中提出了催化劑表面酸堿協(xié)同作用理論,各種助催化劑中,酸性金屬氧化物和堿性金屬氧化物的含量具有特定的比例,但與其絕對(duì)量無關(guān)。據(jù)此原理,調(diào)節(jié)酸性金屬氧化物和堿性金屬氧化物的含量,使催化劑表面酸覆蓋度與堿覆蓋度的比例為最適宜值時(shí),可提高催化劑的活性。通過BET測(cè)定催化劑酸性比表面積和堿性比表面積,并與氨合成活性進(jìn)行關(guān)聯(lián),發(fā)現(xiàn)當(dāng)催化劑表面酸堿覆蓋度的比例為1.1~1.2時(shí),催化劑具有最高的活性。
Almquist等[23]的研究結(jié)果顯示,對(duì)熔鐵催化劑綜合利用堿性和酸性氧化物更為有效,并且兩者的比例因制備方法不同而有不同的影響。Krabetz等[24]研究發(fā)現(xiàn),催化劑的活性與表面的堿度有一定的關(guān)系。劉化章等[25]通過BET表征催化劑酸性比表面積、堿性比表面積與氨合成活性的關(guān)系,發(fā)現(xiàn)當(dāng)催化劑表面酸堿覆蓋度的比例為1.1~1.2時(shí),催化劑具有最高的活性。然而,催化劑表面既要有利于N2的吸附,又要有利于NH3的脫附,因此,調(diào)節(jié)催化劑表面的酸堿性十分必要。
顯然,這種表面酸堿協(xié)同作用與鐵氧化物前驅(qū)體和催化劑制備方法有關(guān)。因此,在鐵氧化物前驅(qū)體為Fe1-xO的前提下,考察了制備方法對(duì)調(diào)節(jié)催化劑表面酸堿性的影響。根據(jù)助催化劑的酸堿性不同,以3種方法加入助催化劑(第1種是不分酸堿性一次性加入助催化劑,第2種是先加酸性助催化劑后加堿性助催化劑,第3種是先加堿性助催化劑后加酸性助催化劑),以考察其對(duì)催化劑氨合成活性的影響,試驗(yàn)結(jié)果如圖3所示。
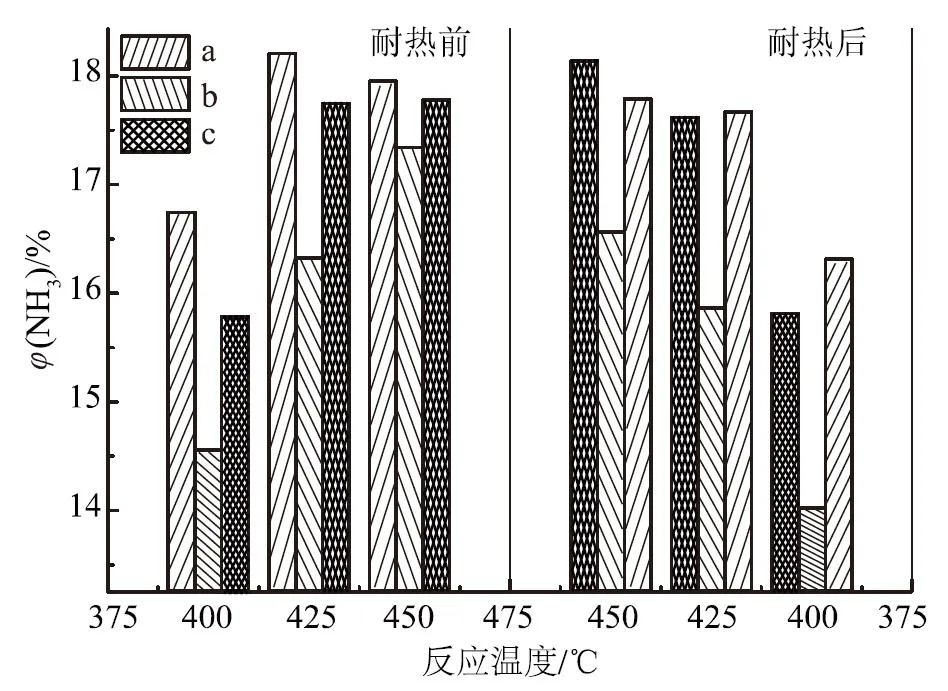
a. 助催化劑一次性加入 b. 分次加入(先加酸性助催化劑) c. 分次加入(先加堿性助催化劑)圖3 助催化劑酸堿性及其加料方式對(duì)催化劑氨合成活性的影響
由圖3可知,助催化劑的酸堿加入順序?qū)Υ呋瘎┌焙铣苫钚杂幸欢ㄓ绊憽T?25 ℃耐熱前,助催化劑一次性加入的催化劑活性比先加堿性助催化劑的高0.45%,比先加酸性助催化劑的高1.87%,說明在催化劑制備過程中,所有助催化劑一次性加入所得的催化劑活性最好,先加堿性助催化劑次之,先加酸性助催化劑的活性最差。比較耐熱前后催化劑氨合成活性可知,在425 ℃時(shí),助催化劑一次性加入的催化劑氨合成活性由耐熱前的18.20%降至耐熱后的17.66%,絕對(duì)值下降0.54%;耐熱后,先加酸性助催化劑的催化劑活性絕對(duì)值下降0.57%;但先加堿性助催化劑的催化劑活性由耐熱前的17.75%降至17.61%,即耐熱前后活性基本不變,說明先加堿性助催化劑能提高催化劑耐熱穩(wěn)定性。由此可知,對(duì)耐熱前的催化劑初活性,助催化劑一次性加入>先加堿性助催化劑>先加酸性助催化劑,助催化劑一次性加入,即傳統(tǒng)的“鍋煮”式熔融法制備的氨合成催化劑具有最佳的活性;但對(duì)于耐熱后的催化劑活性,先加堿性助催化劑>助催化劑一次性加入>先加酸性助催化劑,即先加堿性助催化劑制備的催化劑具有較好的耐熱性,而先加酸性助催化劑的制備方法是不可取的。盡管先加堿性助催化劑制備的催化劑的耐熱性比助催化劑一次性加入略有提高,但由于熔鐵催化劑耐熱性高、使用壽命長,而且分次加入的制備過程較復(fù)雜,所以助催化劑一次性加入,即傳統(tǒng)的“鍋煮”式熔融法仍然是可行的。
上述試驗(yàn)結(jié)果表明,催化劑表面的酸堿性主要是由催化劑前驅(qū)體Fe1-xO決定的,制備方法雖然有一定的影響,但基本上改變不了催化劑表面的酸堿性。
作為一種動(dòng)態(tài)原位分析技術(shù),NH3- TPD技術(shù)目前被廣泛應(yīng)用于催化劑表面酸性的研究。一般來說,不同的脫附峰代表不同類型的活性中心,脫附峰峰頂溫度表征了該中心對(duì)NH3的吸附強(qiáng)度,脫附峰的面積代表該中心活性位數(shù)量。氨分子在催化劑表面存在2種吸附態(tài),即NH3中N- H鍵斷裂的解離吸附和N原子上的孤對(duì)電子與催化劑表面形成的配位吸附。解離吸附為弱吸附,是反應(yīng)進(jìn)行的必要條件;配位吸附為強(qiáng)吸附,由于其占據(jù)了一定活性位,對(duì)反應(yīng)不利。氨合成催化劑的NH3- TPD圖譜如圖4所示。
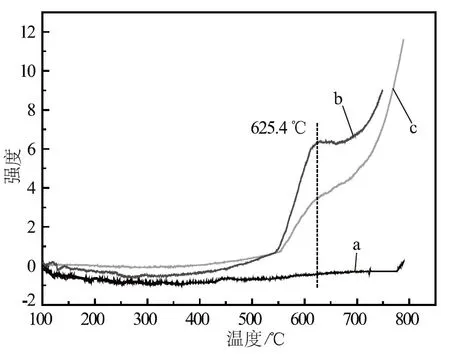
a. 助催化劑一次性加入 b. 分次加入(先加酸性助催化劑) c. 分次加入(先加堿性助催化劑)圖4 催化劑的NH3- TPD圖譜
由圖4可知:在550.0 ℃以下,沒有檢測(cè)到NH3的脫附峰,說明在工業(yè)使用溫度下,催化劑表面呈堿性。在625.4 ℃,助催化劑分次加入的樣品b和樣品c均出現(xiàn)了NH3的脫附峰,這屬于NH3的強(qiáng)吸附位;而助催化劑一次性加入的樣品a在100~800 ℃沒有出現(xiàn)NH3的脫附峰,說明改變助催化劑的加入方式改變了高溫區(qū)的催化劑表面酸性。樣品b表面的酸量高于樣品c,即樣品b表面NH3的強(qiáng)吸附位多于樣品c,那么在氨合成反應(yīng)過程,H2和N2合成的NH3在樣品b表面更難脫附。由于被吸附的氨占據(jù)了催化劑表面的活性位,故樣品b的氨合成活性低于樣品c;而樣品a不存在NH3的強(qiáng)吸附位,故樣品a的氨合成活性最好。
2.3 助催化劑前驅(qū)體及其引入方式對(duì)催化劑活性的影響
催化劑中的氧化鎢除了用于調(diào)節(jié)酸堿性外,還起著增強(qiáng)催化劑對(duì)氮的吸附活化作用。根據(jù)研究氨合成反應(yīng)機(jī)理和鐵催化劑活性本質(zhì)得到的結(jié)論,氮的吸附離解為速控步驟。但在低溫下,催化劑對(duì)氮的吸附能力較弱,而對(duì)氫較強(qiáng),故在低溫下產(chǎn)生了所謂的氫中毒效應(yīng),即H2的強(qiáng)化學(xué)吸附現(xiàn)象。為此,必須添加一些對(duì)氮有較高親和力且熔點(diǎn)比鐵高的金屬或金屬氧化物。本發(fā)明中的鎢有高的親和力,其在氨合成的氮?dú)錃夥罩心鼙徊糠诌€原并與α- Fe形成固溶體,氮以高的鍵能被吸附在鎢上,能明顯增強(qiáng)對(duì)氮的活化,但這又會(huì)使氨合成的第2步——化學(xué)吸附氮的加氫變得困難。但在堿金屬(鉀)存在下,金屬和氮鍵能降低,而且在鎢和鉀的數(shù)量為某確定比例時(shí),其鍵能可以達(dá)到對(duì)于過程的2個(gè)階段以可比的速度進(jìn)行的最佳值。
在熔融法制備催化劑的過程中,存在十分復(fù)雜的化學(xué)反應(yīng),如氧化物前驅(qū)體的分解、固溶體的形成等,所以助催化劑的引入方式不同,制備過程發(fā)生的化學(xué)反應(yīng)就不同,所得催化劑的性能也不同。因此,試驗(yàn)改變助催化劑的前驅(qū)體,如Al2O3和K2O可用KAlO2代替等,這樣制備的氨合成催化劑在性能上有一定的差異。試驗(yàn)設(shè)計(jì)過程主要為:部分助催化劑(Al2O3,K2CO3,CaCO3)在熔融開始前混入熔融爐底部,其他助催化劑在催化劑熔融過程中按CaVO3和CaWO4的混合物、V2O5和WO3的混合物、K2Al2O4,KVO3和K2WO4的混合物3種方式加入。催化劑耐熱前后活性隨溫度變化如圖5所示。
a. 助催化劑一次性加入 b. CaVO3和CaWO4后加 c. V2O5和WO3后加 d. K2Al2O4,KVO3和K2WO4后加圖5 催化劑耐熱前后活性隨溫度變化
由圖5可知:助催化劑分步加入所得催化劑的活性比助催化劑一起加入的高,說明助催化劑分步加入有利于催化劑活性的提高;對(duì)于第2步加入助催化劑的方式,助催化劑化合物的引入方式不同,催化劑的活性也有所不同,此種差異在低溫(425 ℃和400 ℃)下表現(xiàn)得更為明顯;助催化劑釩和鎢以其鈣鹽形式引入比以鉀鹽形式引入所得催化劑的耐熱活性高(耐熱前活性差別較小)。
3 結(jié)語
(1)助催化劑的添加順序?qū)Υ呋瘎┌焙铣苫钚杂幸欢ǖ挠绊憽?/p>
(2)助催化劑按其酸堿性順序加入對(duì)催化劑氨合成活性有一定的影響,其中先加堿性助催化劑制備的催化劑的耐熱穩(wěn)定性高于其他方法制備的催化劑。
(3)助催化劑的分步加入有利于催化劑氨合成活性的提高,并且助催化劑釩鎢以其鈣鹽形式引入比以鉀鹽形式引入所得催化劑的耐熱活性高。
[1] 斯拉克A V,詹姆斯 G R.合成氨:第三冊(cè)[M].北京:化學(xué)工業(yè)出版社,1980.
[2] FERTIMONT S P A. Process for preparing iron- based catalysts for the synthesis of ammonia and catalysts so obtained:US4789657A[P].1988- 12- 06.
[3] UNIV BOSTON. Partially reduced ferric oxide catalyst for the making of ammonia via the photoassisted reduction of molecular nitrogen and method for the preparation of the catalyst:US4703030A[P].1987- 10- 27.
[4] KLISSURSKI D G, MITOV I G, TOMOV T. A Study Of The Preparation And Properties Of Precipitated Iron Catalysts For Ammonia Synthesis[J]. Studies in Surface Science and Catalysis,1983,16:421- 430.
[5] IMPERIAL CHEMICAL INDUSTRIES PLC. Iron catalyst and method of producing it:US4668658A[P].1987- 05- 26.
[6] MINI RICERCA TECNOLOG. Process for producing the precursor of a precipitated catalyst for the ammonia synthesis:EP0459424A1[P].1991- 12- 04.
[7] ESUEIRAS J, HOMS N, PISCINA P D L, et al. Structure and reactivity of alumina- supported iron catalysts for ammonia synth[J]. Journal of Catalysis,1986(2):264- 276.
[9] SANTOS J, PHILLIPS J, DUMESIC J A. Metal- support interactions between iron and titania for catalysts prepared by thermal decomposition of iron pentacarbonyl and by impregnation[J]. Journal of Catalysis,1983(1):147- 167.
[10] 劉化章.氨合成催化劑——實(shí)踐與理論[M].北京:化學(xué)工業(yè)出版社,2007.
[11] LIU Huazhang. Ammonia synthesis catalyst 100 years: Practice, enlightenment and challenge[J]. Chinese Journal of Catalysis,2014(10):1 619- 1 640.
[12] 劉化章,李小年,胡樟能,等.Fe1-xO基氨合成催化劑的制備化學(xué)[J].高等學(xué)校化學(xué)學(xué)報(bào),2002(1):87- 91.
[13] 張?zhí)烀鳎聊埽瑒⒒?制備條件對(duì)FeO基氨合成催化劑性能的影響[J].化學(xué)通報(bào)(網(wǎng)絡(luò)版),2003,66:w81.
[14] 劉化章,李小年.Fe1-xO基氨合成催化劑高活性機(jī)理初探[J].催化學(xué)報(bào),2005(1):79- 86.
[15] 劉化章,李小年.熔鐵催化劑活性與其母體鐵氧化物形態(tài)和組成的關(guān)系(I)母體鐵氧化物活性次序[J].化工學(xué)報(bào),1998(5):534- 541.
[18] 劉化章,李小年,鈴木聰雄,等.熔鐵催化劑活性與其母體鐵氧化物形態(tài)和組成的關(guān)系(II)表面活性位和氨合成反應(yīng)速率[J].化工學(xué)報(bào),2000(4):462- 467.
[19] 李小年,傅冠平,劉化章,等.助催化劑對(duì)Fe1-xO基氨合成催化劑還原性能的影響[J].催化學(xué)報(bào),1998(1):24- 28.
[20] 李小年,劉化章,陳誦英.助催化劑對(duì)Fe1-xO基氨合成催化劑活性的影響[J].催化學(xué)報(bào),1998(3):201- 205.
[21] 李小年,管升,劉化章,等.新體系氨合成催化劑母體中Fe1-xO的歧化及其效應(yīng)[J].催化學(xué)報(bào),1998(4):315- 319.
[22] 李小年,劉化章,許裕生,等.助催化劑對(duì)氨合成Fe1-xO基催化劑母體歧化反應(yīng)的抑制作用的穆斯堡爾譜研究[J].催化學(xué)報(bào),1999(1):76- 80.
[23] ALMQUIST J A, CRITTENDEN E D. A Study of Pure- Iron and Promoted- Iron Catalysts for Ammonia Synthesis1[J]. Industrial & Engineering Chemistry,1926(12):1 307- 1 309.
[24] KRABETZ R, PETERS C. The Function of the promotors in the Technical Ammonia Catalyst[J]. Angewandte Chemie International Edition,1965(4):341- 347.
[25] 劉化章,鈴木聰雄,大西龍一郎.母體鐵氧化物對(duì)鐵催化劑表面性質(zhì)和活性的影響[C]∥第九屆全國催化學(xué)術(shù)會(huì)議論文集.北京:海潮出版社,1998.
Study of Preparation of Fe1-xO Based Ammonia Synthesis Catalyst by Melting Method Feeding by Stages
HUANG Shiliang, CHENG Tianhong, HAN Wenfeng, LIU Huazhang
(Institute of Industrial Catalysis, Zhejiang University of Technology Zhejiang Hangzhou 310032)
The Fe1-xO based ammonia synthesis catalyst is prepared with melting method, based on properties of oxide cocatalysts including melting point, crystal form and acid- base properties, the effects of feeding method of adding cocatalyst in sequence and by stages during catalyst preparation on catalyst activity and thermal stability are investigated. Results show that adding order of cocatalyst has a certain effect on activity of ammonia synthesis catalyst; adding cocatalysts according to their acid- base property sequence have effects on activity of ammonia synthesis catalyst, the thermal stability of the catalyst, which is prepared by adding alkaline cocatalyst first, is higher than that of other catalysts prepared with other methods; The catalyst which cocatylists vanadium and tungsten are introduced in calcium salt form has higher thermal stability than that catalyst which cocatalyst is introduced in form of potassium salt.
melting method ammonia synthesis Fe1-xO based catalyst preparation method
黃仕良(1988—),男,碩士,從事氨合成催化劑研究。
韓文鋒,男,副研究員,從事催化劑工程研究;hanwf@zjut.edu.cn。 劉化章,男,研究員,從事催化劑工程研究;cuihua@zjut.edu.cn。
TQ426.6
A
1006- 7779(2016)06- 0019- 06
2015- 11- 27)
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時(shí)代(2018年3期)2018-06-11 16:10:44
浙江大學(xué)學(xué)報(bào)(工學(xué)版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(xué)(2015年4期)2016-01-17 09:01:27
應(yīng)用化工(2014年3期)2014-08-16 13:23:50
