以發熱、皮疹、血常規兩系減少為表現的噬血細胞綜合征1例
2016-06-16 08:26:12遲磊黃燕崔振澤
中國中西醫結合兒科學 2016年2期
關鍵詞:兒童
遲磊, 黃燕, 崔振澤
116012 遼寧 大連,大連市兒童醫院呼吸內科
?
病例報告
以發熱、皮疹、血常規兩系減少為表現的噬血細胞綜合征1例
遲磊,黃燕,崔振澤
116012 遼寧 大連,大連市兒童醫院呼吸內科
【關鍵詞】噬血細胞綜合征;發熱;皮疹;血常規兩系減少;兒童
通過對1例病程短、進展快、病情兇險的“噬血細胞綜合征”患兒的診斷和搶救過程的討論,對噬血細胞綜合征的病因、流行病學、臨床表現、實驗室檢查、診斷、治療和預后進行了系統綜述,總結出經驗教訓供兒科醫生參考。
1病情摘要
1.1現病史患兒,女,3.5歲,因“發熱4 d、皮疹及出血點2 d”于2012年9月5日13:00入院,4 d前無明顯誘因出現發熱,體溫最高39 ℃,無寒戰抽搐,無咳不喘,非噴射性嘔吐胃內容物2次,無膽汁及咖啡樣物,排糊樣便,每日1~2次,無黏液膿血。2 d前發現周身皮疹及雙下肢出血點,于門診輸液“美洛西林”1次(劑量不詳),皮疹逐漸增多,精神不振,今日來診,傳染科門診除外“猩紅熱”,皮膚科門診考慮“多形性紅斑”,建議內科抗感染治療觀察,內科門診以“發熱待查:血液系統疾病待除外”收入院,病來精神食欲欠佳,睡眠偏多,尿略少。
1.2既往史體健,家中養狗,有犬類密切接觸史,無藥物過敏史,無手術外傷輸血史,無傳染病接觸史。
1.3家族史姐姐年幼時曾患過同樣“發熱、皮疹、出血點”疾病,服中藥后痊愈。
1.4體格檢查體溫36.3 ℃,脈搏100次/分,呼吸20次/分,體質量15 kg,血壓84/50 mm Hg(1 mm Hg=0.133 kPa),神志清楚,精神不振,呼吸尚平穩,顏面及周身皮膚潮紅,可見多量大小不等紅斑,略突出于體表,部分融合成片,雙小腿脛側可見散在針尖大小出血點,未見明顯瘀斑,雙眼球結膜充血,雙眼瞼及周身無明顯水腫,頸部可觸及2~3枚腫大淋巴結,最大1.5 cm×2.0 cm,質中,活動可,無觸痛,咽充血,頸軟,雙肺呼吸音粗,心音有力,律齊,無雜音,腹軟,肝肋下剛及,脾未及,腸鳴音正常,肢端溫,神經系統未見異常體征。
1.5輔助檢查血常規(2012年9月5日門診):0.48×109/L,中性粒細胞0.14,血紅蛋白113.3 g/L,血小板計數39×109/L。尿常規(2012年9月5日門診):尿混濁,有沉淀物,尿蛋白(++),紅細胞0~1個/HP,白細胞3~5個/HP。
1.6臨床初步診斷發熱及二系減少原因待查。
1.7診療過程入院后即查頭顱CT,未見明顯異常。給予磷霉素鈉、炎琥寧抗感染,維生素C及爐甘石洗劑減輕皮疹,對癥支持治療。
患兒入院后仍有發熱,精神萎靡,易激惹,不進食,四肢末梢涼,急查動脈血氣、出凝血篩查、生化全項。20:00給予止血敏、止血芳酸、維生素K1、止血合劑治療,復查血常規、出凝血篩查,給予新鮮冰凍血漿150 mL及血小板1份。23:45患兒嘔吐咖啡樣物2次,四肢末梢涼,測血壓2次均下降,無尿,于9月6日0:50轉入ICU病房搶救,下胃腸減壓管吸出鮮紅色胃內容物,給予擴容、繼續輸血漿、血小板等對癥支持治療,復查動脈血氣分析、血常規、出凝血篩查、生化全項,輸血后患兒病情稍平穩,于9月6日8:00,家長簽字帶患兒離院,前往上級醫院進一步診治。
各項檢測指標分別見表1~4。
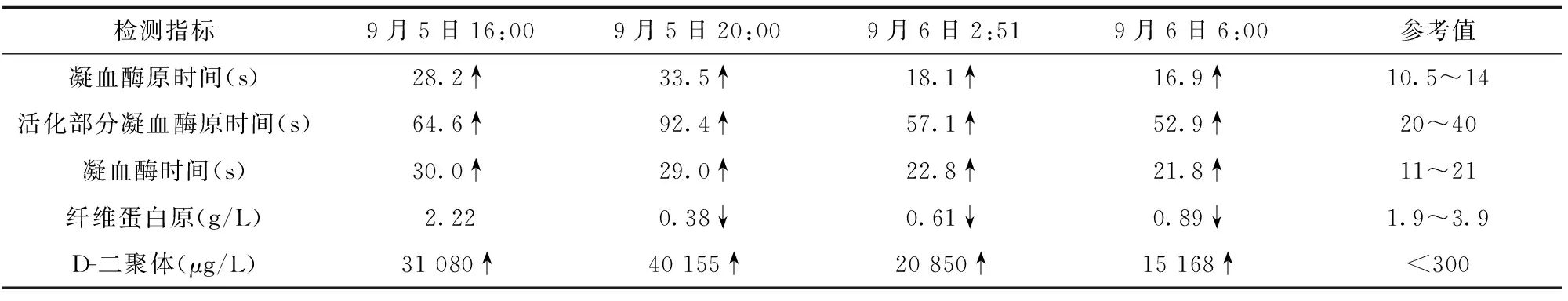
表1 出凝血篩查結果

表2 生化全項結果
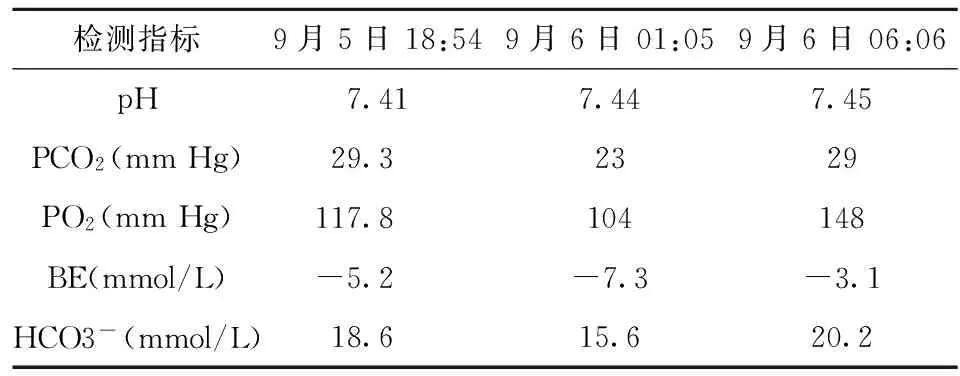
表3 動脈血氣分析結果
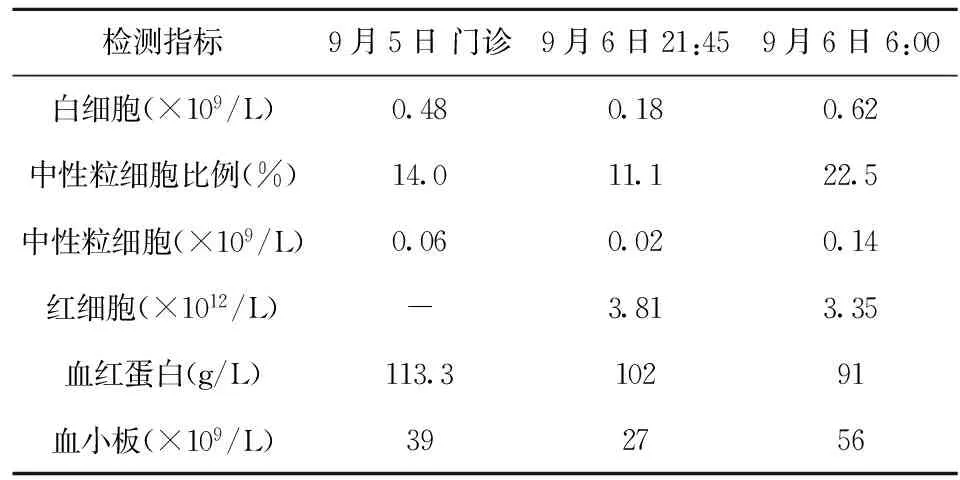
表4 血常規結果
2臨床病例討論(按接診順序討論)
2.1住院醫師一(血液病房首診醫生)該患兒于9月5日13:00入本科病房,發熱4 d,皮膚可見皮疹及出血點,精神欠佳,血常規二系減少,初步診斷“發熱及二系減少原因待查”,首先考慮感染所致的二系減少:(1)原蟲感染:患兒發熱、吐瀉、精神食欲不振、周身可見皮疹,有犬類密切接觸史,粒系及血小板減少,尿含蛋白并可見白細胞,需考慮“斑疹傷寒”等原蟲感染性疾病;(2)泌尿系感染:患兒尿常規異常并伴有發熱,需考慮尿路感染,應留取中段尿培養檢查;(3)呼吸道感染:患兒發熱、咽充血、雙肺呼吸音粗,不除外呼吸道感染,需拍胸片助診。
2.2住院醫師二(血液病房夜班醫生)本患兒除了感染所致的二系減少,還要考慮非感染性疾病:(1)血液系統疾病:發熱、淋巴結腫大,二系減少,必須做骨髓穿刺除外血液系統疾病;(2)朗格罕綜合征:發熱、皮疹、淋巴結腫大、二系減少,同樣需做骨髓穿刺除外;(3)結締組織疾病:發熱、皮疹、粒系及血小板減少,結締組織疾病需要考慮,進一步完善相關檢查助診。
2.3副主任醫師(醫療總值班)基本同意兩位醫生的初步診斷意見,可以逐步完善上述相關檢查進一步確診。目前患兒的特點是病程短、進展快、精神不振,入院后煩躁與嗜睡交替,易激惹,心、肺、腹、神經系統查體未見明顯異常,凝血酶原時間、活化部分凝血活酶原時間、凝血酶時間均延長、D-二聚體顯著升高,提示一個消耗性低凝狀態,需立刻完善頭顱CT,明確有無顱內出血,同時動態監測血小板計數變化,復查出凝血篩查了解凝血功能狀態,急查生化全項了解肝腎功能狀態,密切觀察皮膚、齒齦、鼻腔、穿刺點的出血傾向及出血程度,動態監測血壓,準備處理可能出現的休克及微循環障礙。診斷首先還是考慮嚴重感染所致可能性大,立即采血培養除外膿毒血癥,給予有效抗生素積極控制感染。至于斑疹傷寒,與環境和個人衛生條件密切相關,隨著衛生環境的改善,這類疾病發病率逐年降低,但仍需追問患兒居住環境有無鼠患,有無野外草叢隨意坐臥或農作物接觸史。
2.4主治醫師(內科總住院醫師)20:00第二次“出凝血篩查”回報,凝血酶原時間、活化部分凝血活酶原時間、凝血酶時間仍延長、D-二聚體仍顯著升高,纖維蛋白原降低,“生化全項”回報提示甘油三酯、血清鐵蛋白、β2微球蛋白、膽紅素、轉氨酶、乳酸脫氫酶、膽汁酸、肌酸激酶同工酶、尿素氮、C反應蛋白等均明顯升高,電解質K+、Na+、Cl-、Ca2+均降低,提示病情的復雜性及嚴重性,但多臟器受累程度與臨床表現似乎并不相符,患兒目前生命體征尚平穩,無明顯休克征象,診斷比較困難,治療仍以對癥為主,建議給予止血敏、止血芳酸、維生素K1、止血合劑治療,暫可不必給予肝素抗凝,密切監測出凝血功能變化及生命體征。
2.5住院醫師三(ICU病房夜班醫生)患兒發熱、嘔吐、精神不振、煩躁嗜睡交替、查體無神經系統的局灶體征,血清轉氨酶升高、肝功能異常而無黃疸、血糖低、凝血酶原時間延長,是否要考慮“瑞氏綜合征”?應該詢問一下近期感染史與用藥史、可疑中毒史。
2.6副主任醫師(醫療總值班)“瑞氏綜合征”給予一個非常好的提醒,經再次詢問家長,患兒發病前2周內并無呼吸道或消化道感染的前驅癥狀,也無明確的特殊藥物毒物接觸史,目前有幾點不支持“瑞氏綜合征”:中樞神經系統受累表現并不典型,無進行性的腦部癥狀、肝臟不大、白細胞不高、膽紅素升高(瑞氏綜合征不影響膽紅素代謝)、頭顱影像并無彌漫性或局部性水腫的表現,最重要的檢查血氨目前夜間無法檢測,明日應送檢,必要時行腦脊液檢查以助鑒別診斷。
2.7主任醫師(血液病房)患兒病情持續進展,血壓降低、四肢涼、嘔吐咖啡樣物,復查血常規及出凝血篩查繼續惡化,綜合其臨床特點:病程短、進展快、病情兇險、外周血常規兩系減少合并甘油三酯、膽紅素、血清鐵蛋白、乳酸脫氫酶增高伴低鈉血癥,迅速出現凝血功能障礙(低纖維蛋白原血癥、凝血酶原時間延長、活化部分凝血活酶時間延長)以致早期彌漫性血管內凝血征象等,基本符合“噬血細胞綜合征”的臨床診斷標準,確診需反復骨穿尋找吞噬性組織細胞,目前建議給予血漿、血小板等對癥支持治療,該病兒科罕見,癥狀兇險,死亡率高,應向家長充分交代,待病情稍平穩后轉入上級醫院繼續治療。
此患兒最終診斷“噬血細胞性淋巴組織細胞增生癥”(hemophagocytic lymphohistiocytosis,HLH),亦稱“噬血細胞綜合征”,是一組以在骨髓或其他淋巴組織/器官中出現異常增多的組織細胞且伴有吞噬自身血細胞行為為特征的病癥,其特點是發熱、肝脾腫大、全血細胞減少、骨髓、肝、脾、淋巴結中噬血性組織細胞增多以及高甘油三酯血癥、低纖維蛋白原血癥、高細胞因子血癥等。由于該病特異性檢查指標少,目前尚無統一的診斷標準,因此在臨床診治中存在一定困難,正確認識該病的臨床特點、實驗室檢查、干預措施是搶救成功的關鍵。
3發病率和流行病學
兒童HLH分為原發性和繼發性兩種。原發性者發病率每年約1/10萬[1],繼發性HLH的確切發病率尚不得而知,患兒多存在與免疫缺陷或免疫抑制治療有關的原發疾病,致病病原體以EB病毒感染最為常見[2],其他包括人類皰疹病毒6型、巨細胞病毒、腺病毒、水痘-帶狀皰疹病毒、單純皰疹病毒、寇熱立克次體[3]和麻疹病毒及細菌、真菌、原蟲等[4-5]。
4癥狀和體征
(1)典型表現[6]:間斷或持續發熱、肝脾顯著腫大、血細胞減少,血小板減少時可出現紫癜和出血;(2)中樞神經系統受累多發生在病程的晚期,出現進行性的腦和腦膜癥狀,包括易激惹、前囟膨出、頸項強直、張力減低或增高和驚厥等;(3)一過性皮疹,疹型無特異性,常與高熱并行;(4)半數患者可有淋巴結腫大;(5)淋巴細胞和巨噬細胞聚集于肺部時可出現相應癥狀,容易與肺部感染混淆;(6)非特異性癥狀:如蒼白、浮腫、黃疸、食欲減低和生長遲緩等。
5實驗室檢查
(1)血細胞減少:以血小板減少和貧血最常見,白細胞減少較輕。血小板的變化可能預示疾病的活動程度。(2)高甘油三酯血癥,極低密度脂蛋白和低密度脂蛋白明顯增高,高密度脂蛋白減低;低纖維蛋白原血癥常見,凝血酶原時間延長,活化部分凝血活酶時間延長;肝功異常[6]可表現為轉氨酶增高或膽紅素增高,鐵蛋白升高[7]、低鈉血癥和白蛋白/總蛋白比值降低亦較常見。(3)腦脊液檢查:淋巴細胞增多[(5~50)×106/L],可見單核細胞,但噬血細胞少見[8],伴蛋白質增高。(4)影像學檢查:肺部受累可見中度間質浸潤。MR和CT可發現腦部炎癥或脫髓鞘病變,也可發現腦出血、腦萎縮和腦水腫。(5)骨髓象:疾病早期噬血現象并不明顯,僅表現為中等度的骨髓增生,可見反應性的組織細胞增生,無惡性細胞浸潤;極期除組織細胞增多外,可見或多或少的噬血細胞,主要吞噬紅細胞,也可吞噬血小板及有核細胞;晚期骨髓增生度降低。診斷不明確時,應連續多次查骨髓象,以便早期發現吞噬現象。
6診斷
HLH缺乏特異性的實驗室診斷方法,診斷較為困難,誤診和漏診率高。依據HLH-2004方案,以下8條有5條符合即可診斷HLH[6]。(1)發熱;(2)脾臟增大;(3)外周血至少兩系減少,血紅蛋白<90 g/L,血小板<100×109/L,中性粒細胞<1.0×109/L;(4)高甘油三酯血癥和(或)低纖維蛋白原血癥;(5)骨髓、脾臟或淋巴結中有噬血現象;(6)自然殺傷細胞活力降低或缺乏;(7)血清鐵蛋白≥500 mg/L;(8)可溶性CD25≥2 400 U/mL。同時,血清或尿中的β2微球蛋白可反映疾病的活動度;所有患者在起病時均應進行自然殺傷細胞活性檢查。
有些病例的表現并非與上述標準完全一致。有些起病時脾大并不顯著,起病時也并非總能找到噬血細胞,連續做骨髓檢查可能有助于診斷,如肝臟的組織學所見類似于慢性持續性肝炎、腦脊液中見單個核細胞增多亦支持HLH的診斷。
7治療和預后
噬血細胞綜合征臨床過程兇險,病死率高,早期診斷、早期治療是搶救成功的關鍵,通過本例患兒的搶救過程,筆者深刻認識到,一旦臨床高度懷疑本病,應積極干預治療,及早給予大劑量激素[6]聯合替代療法(補充血漿、紅細胞或血小板),同時給予大劑量丙種球蛋白,對減少造血負調控類的細胞因子產生和減弱巨噬細胞的吞噬活性有一定的作用,不能因等待骨髓象找到噬血細胞而耽誤臨床治療。一旦骨髓象確診后,早期給予以下治療可能會改變疾病的進程:(1)化學療法:常用的化療藥物有細胞毒性藥物;(2)免疫治療:環胞菌素A[5]/抗胸腺細胞球蛋白可誘導緩解;(3)造血干細胞移植[9-10]。
參考文獻
[1]Niece JA,Rogers ZR,Ahmad N,et al.Hemophagocytic lymphohistiocytosis in Texas:observations on ethnicity and race[J].Pediatr Blood Cancer,2010,54(3):424-428.
[2]Przybylski M,Dzieciatkowski T,Zduńczyk D,et al.Microbiological findings and treatment of EBV-associated hemophagocytic lymphohistiocytosis:a case report[J].Arch Immunol Ther Exp(Warsz),2010,58(3):247-252.
[3]Lecronier M,Prendki V,Gerin M,et al.Q fever and Mediterranean spotted fever associated with hemophagocytic syndrome:case study and literature review[J].Int J Infect Dis,2013,17(8):e629-633.
[4]Janka G.Hemophagocytic lymphohistiocytosis:when the immune system runs amok[J].Klin Padiatr,2009,221(5):278-285.
[5]Rouphael NG,Talati NJ,Vaughan C,et al.Infections associated with haemophagocytic syndrome[J].Lancet Infect Dis,2007,7(12):814-822.
[6]Henter JI,Horne A,Aricó M,et al.HLH-2004:Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis[J].Pediatr Blood Cancer,2007,48(2):124-131.
[7]Allen CE,Yu X,Kozinetz CA,et al.Highly elevated ferritin levels and the diagnosis of hemophagocytic lymphohistiocytosis[J].Pediatr Blood Cancer,2008,50(6):1227-1235.
[8]Janka GE.Familial and acquired hemophagocytic lymphohistiocytosis[J].Annu Rev Med,2012,63:233-246.
[9]Cooper N,Rao K,Gilmour K,et al.Stem cell transplantation with reduced-intensity conditioning for hemophagocytic lymphohistiocytosis[J].Blood,2006,107(3):1233-1236.
[10]Worth AJ,Nikolajeva O,Chiesa R,et al.Successful stem cell transplant with antibody-based conditioning for XIAP deficiency with refractory hemophagocytic lymphohistiocytosis[J].Blood,2013,121(24):4966-4968.
作者簡介:遲磊(1976-),女,醫學碩士,副主任醫師。研究方向:小兒呼吸內科疾病的診治通訊作者:黃燕,E-mail:sunnyyanzi@126.com
doi:10.3969/j.issn.1674-3865.2016.02.041
【中圖分類號】R725
【文獻標識碼】B
【文章編號】1674-3865(2016)02-0245-04
(收稿日期:2015-08-19)(本文編輯:劉穎)
猜你喜歡
少兒美術·書法版(2021年12期)2021-10-24 02:50:16
少兒美術·書法版(2021年9期)2021-10-20 06:35:28
少兒美術·書法版(2021年7期)2021-10-20 06:29:16
少兒美術·書法版(2021年11期)2021-10-20 06:23:28
少兒美術·書法版(2021年10期)2021-10-20 06:14:04
少兒美術·書法版(2021年8期)2021-10-20 06:08:10
少兒美術(2019年8期)2019-12-14 08:07:00
少兒美術(2019年3期)2019-12-14 08:02:56
雜文選刊(2016年7期)2016-08-02 08:39:56
小天使·一年級語數英綜合(2016年6期)2016-05-14 12:21:05
