頭孢呋辛酯的合成研究
2016-07-18 05:29:17曹衛凱
合成技術及應用 2016年2期
曹衛凱
(西安萬隆制藥股份有限公司,陜西西安 710119)
?
研究論文
頭孢呋辛酯的合成研究
曹衛凱
(西安萬隆制藥股份有限公司,陜西西安710119)
摘要:利用7-氨基頭孢烷酸(7-ACA)合成頭孢呋辛酯,同時對合成過程及關鍵點進行優化。首先7-ACA在甲醇和水的混合溶劑中低溫水解,生成D-7-ACA,不經分離,直接進行7-位酰化反應,經結晶分離干燥后得到關鍵中間體去氨甲酰基頭孢呋辛酸(DCC),收率可達84.8%,再經過與氯磺酸異氰酸酯(CSI)反應完成C-3位的氨甲酰化改造,得到頭孢呋辛酸的溶液,結晶分離干燥后,收率可達91.4%,最后與1-溴乙基乙酸酯縮合得到頭孢呋辛酯,收率可達93.8%,總合計收率達72.7%。該工藝具有高收率、低成本、易操作等特點,極具有工業化生產價值。
關鍵詞:頭孢呋辛酸頭孢呋辛酯合成工業化
頭孢呋辛酯是頭孢呋辛(API)的前體藥物,屬于半合成的第二代廣譜頭孢菌素類藥物,是英國葛蘭素史克(GSK)公司開發的頭孢菌素類抗生素,本品抗菌譜廣,對多種革蘭氏陽性和革蘭氏陰性細菌有效,同時對β-內酰胺酶穩定,由于其療效確切,腎臟毒性低、副作用小[1-2],被廣泛應用于對抗敏感菌引起的各類感染。其抗菌作用機制是通過結合細菌蛋白,抑制細菌細胞壁的合成,使細菌細胞壁破壞缺陷,菌體內物質外漏,致菌體死亡[3-5]。臨床使用頭孢呋辛酯主要用于治療呼吸道及耳鼻喉、皮膚軟組織及泌尿生殖系統的感染,同時對敗血癥、腦膜炎及骨關節感染也有很好的療效。頭孢呋辛酯己成為抗感染藥物中的一線用藥,同時為國家基本藥物,自2009年9月21日開始施行《國家基本藥物目錄》以來,頭孢呋辛酯一直被收錄其中。
頭孢呋辛酯的合成是經過將頭孢呋辛酸的羧基進行成酯改造而得,一般都是直接與1-溴乙基乙酸酯直接反應,得到頭孢呋辛酯[6],而頭孢呋辛酸合成路線有多條,主要的工藝路線有以下幾種:1)以7-ACA為起始物,溶解后,用甲氧亞胺基呋喃乙酰氯進行7位氨基酰化,之后3位水解,得到3-去氨甲酰基-頭孢呋辛酸,再將3位羥甲基改造得到頭孢呋辛酸[7-9];2)以7-ACA為起始物,水解得3-去乙酰基-7-氨基-頭孢烷酸(D-7-ACA),然后將3位的羥甲基改造為氨甲酰氧甲基,再將7位氨基酰化引入側鏈甲氧亞胺基呋喃乙酸銨鹽(SMIA)得到頭孢呋辛酸[10];3)以7-ACA為起始物,水解得D-7-ACA,先進行7位氨基的酰化將側鏈引入,得到3-去氨甲酰基-頭孢呋辛酸,再用氯磺酰異氰酸酯進行3位的羥甲基改造,同樣可得頭孢呋辛酸[11-17]。其中方法1,由于先進行7位氨基改造,這樣使得在水解3位時,7位酰胺鍵也發生斷裂,產生雜質D-7-ACA,使得最終產品純度大大降低;方法2雖然先進行了3位酯鍵(R-CH2-O-COCH3)的水解,但之后直接進行3位羥甲基(R-CH2-OH)改造,得到氨甲酰氧甲基取代物(R-CH2-O-CONH2),這樣會使得在進行7位氨基改造時,3位裸露的氨基同樣會進行反應,產生3位氨基的酰化雜質(R-CH2-O-CONH-R1),R1為甲氧亞胺基呋喃乙酰。
為了制備高純度的頭孢呋辛酯,本研究以方法3為基礎,同時對合成過程關鍵工藝參數進行優化,提高成品質量,同時將其收率大大提高,經過筆者優化后的頭孢呋辛酯合成工藝收率,期望遠高于已有的報道合成方法[6,9]。
1試驗
1.1主要合成原料與試劑
7氨基頭孢烷酸7-ACA,純度≥98.5%,武漢楷倫;甲氧亞胺基呋喃乙酸銨鹽SMIA,純度≥99.0%,江蘇清泉;氯磺酰異氰酸酯CSI,工業級,四平精細化工;1-溴乙基乙酸酯,工業級,四平精細化工。
三氯氧磷、DMF、甲醇、乙腈、乙酸乙酯、異丙醚均為分析純,均為國藥集團化學試劑有限公司制;碳酸氫鉀,分析純,為天津福晨化學試劑廠制;活性炭,徐州天正活性炭廠制。
1.2主要儀器設備
酸度計,梅特勒FE20K Plus型;低溫循環泵,長城科工貿DLSB-30/-40型;自動水分測定儀,先驅威鋒ZDJ-3S型;高效液相色譜儀,島津LC-2010 CHT型。
1.3實驗過程
具體合成工藝為:以7-ACA水解得到D-7-ACA,產物不經分離,直接在水相中與7位側鏈酰氯進行混合相反應,制得3-去氨甲酰基-頭孢呋辛酸(DCC),DCC經分離、干燥后與CSI反應,水解得到頭孢呋辛酸溶液,之后進行結晶、過濾、干燥后可得頭孢呋辛酸成品,將頭孢呋辛酸溶解成鹽,成鹽后不經過分離,直接加入1-溴乙基乙酸酯成酯,經過萃取、脫色處理,之后用析晶溶劑析晶,最后過濾洗滌,減壓干燥得到頭孢呋辛酯成品,如圖1所示。
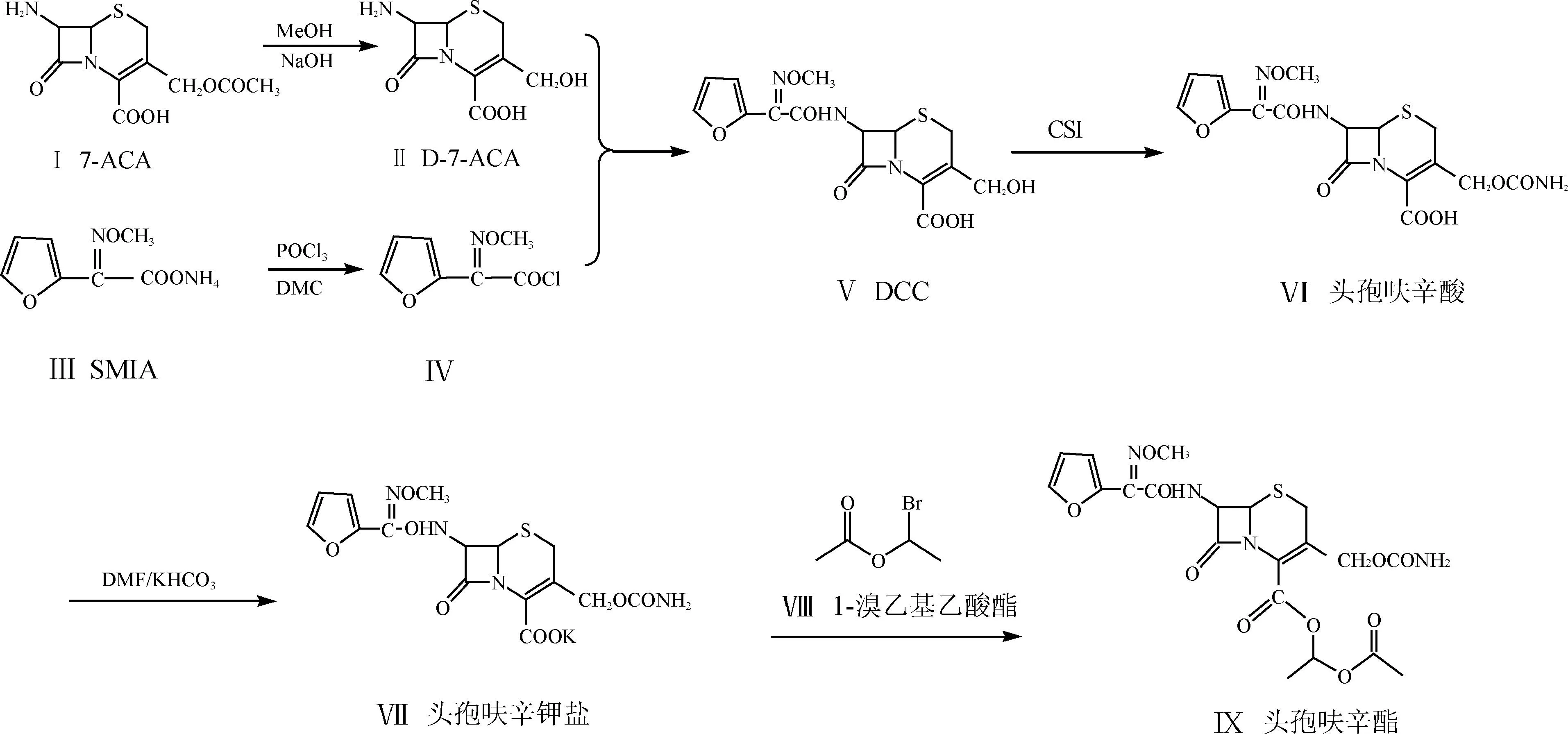
圖1 頭孢呋辛酯的合成路線
1.3.1甲氧亞胺基呋喃乙酰氯(Ⅳ)的合成
在250 mL三口反應瓶中加入DMF(30 g,0.41 mol)、DMAC(36 g,0.41 mol)和Ⅲ SMIA(17 g,0.091 mol),之后再加入DMC(40 mL),在攪拌下降溫到-20 ℃,加入三氯氧磷(16.9 g,0.110 mol),控溫-15~-10℃,反應120 min,得含Ⅳ反應液,降溫至(-30±5)℃備用。
1.3.23-去氨甲酰基-頭孢呋辛酸DCC(Ⅴ)的合成
在500 mL三口反應瓶中加入甲醇(120 mL)、水(80 mL)和Ⅰ7-ACA(24 g,0.088 mol),攪拌下降溫至-35 ℃,滴加10%氫氧化鈉(60 mL),攪拌30 min后,在25 min內滴加10%鹽酸(20 mL),加入備用酰氯Ⅳ,反應30 min后,加入DMC(90 mL)進行洗滌,向水相中滴加10%鹽酸,調pH到2.0,過濾,濾出固體用水洗滌,真空干燥得淡黃色粉末DCC(28.5 g,0.074 mol,收率84.8%,水分0.41%)
1.3.3頭孢呋辛酸(Ⅵ)的合成
在500 mL三口瓶反應中加入乙腈(97 mL),啟動攪拌降溫到-30 ℃,加入中間體DCC(28.5 g,0.074 mol),控溫至-30~-20 ℃,加入CSI(15 g,0.106 mol),保溫-28~-23 ℃,反應80 min,加入水(40 mL),反應180 min,再向反應液中加入水(200 mL),控溫15~25 ℃,滴加氨水,在45 min內,調節pH到5.6,加入DMC(20 mL)萃取分層,向水層中加入10%鹽酸,控制攪拌轉速,調pH到2.0,過濾,濾出固體用水洗滌,真空干燥得頭孢呋辛酸(29 g,0.068 mol,水分0.26%,收率91.4%)。
1.3.4頭孢呋辛酯(Ⅸ)的合成
在500 mL反應瓶中加入DMF 100 mL,控制料液溫度25~30 ℃,快速攪拌下投入碳酸氫鉀12 g,緩慢加入頭孢呋辛酸(20 g,0.047 mol),控制料液溫度25~35 ℃,攪拌60 min,反應完成后,控制料液溫度-15~-10 ℃,然后快速將預冷至-15℃下的1-溴乙基乙酸酯Ⅷ(11.8 g,0.071 mol)加入到混合液中,該過程控溫≤-10 ℃,控制料液溫度-15~-10 ℃,繼續攪拌反應30~40 min,將酯化反應液加入到冷卻至5 ℃以下的乙酸乙酯600 mL與1.0%NaHSO3溶液200 mL混合溶液中,控制料液溫度5~10 ℃,攪拌5 min,靜置15 min,收集有機層,將預冷3.0%的鹽酸溶液600 mL滴加到有機層中,攪拌5 min,靜置30 min,收集水層,水層用乙酸乙酯200 mL洗滌,洗滌后的水層加入活性炭1.0 g,15~25 ℃攪拌脫色30 min,過濾,控制料液溫度0~10 ℃,攪拌下加入冷二氯甲烷400 mL,控制料液溫度0~10 ℃,用5%的氨水調節溶液的PH 8.0~8.5,靜置20 min,收集有機層,有機層用2.0%的NaCl溶液500 mL洗滌,硫酸鈉干燥有機層,減壓濃縮至總體積的30%,控制料液溫度0~10 ℃,將冷異丙醚500 mL在30 min內加入到濃縮液中,緩慢攪拌養晶,得頭孢呋辛酯的混懸液,過濾,用異丙醚適量洗滌,抽干,得頭孢呋辛酯濕品,40 ℃下真空干燥,得頭孢呋辛酯Ⅸ(22.5 g,0.044 mol),收率93.8%,水分:0.52%,產品純度為99.4%(HPLC),1H-NMR(DMSO-d6)δ:9.70(m,1H),7.82(d,1H,J=1.78 MHz),6.90(m,1 H,J=5.33 MHz),6.62(m,1 H,J=1.78 MHz),6.55(m,1 H,J=1.78 MHz,J=3.52 MHz),5.82~6.76(br,2H),5.50(m,1H),5.32(m,1H),4.70(m,2H),3.95(s,3H),3.60~3.82(m, 2H),2.06(d,3H),1.52(m,3H,J=5.72 MHz).
1.4質量分析
該實驗過程合成所得頭孢呋辛酯成品純度高,尤其在最后頭孢呋辛酯合成步驟,增加了相轉移過程,使目標產物先從有機相中轉移到水相中,再從水相轉移到有機相中,每一次轉移都用相應溶劑進行洗滌,最后再在有機相中通過析晶溶劑將其析出,這樣能使反應產生雜質有效被除掉,最終通過相轉移萃取洗滌結晶出來的產品純度比直接析晶出來產品純度高,具體參數見下表1。
表1相轉移反應對頭孢呋辛酯質量的影響
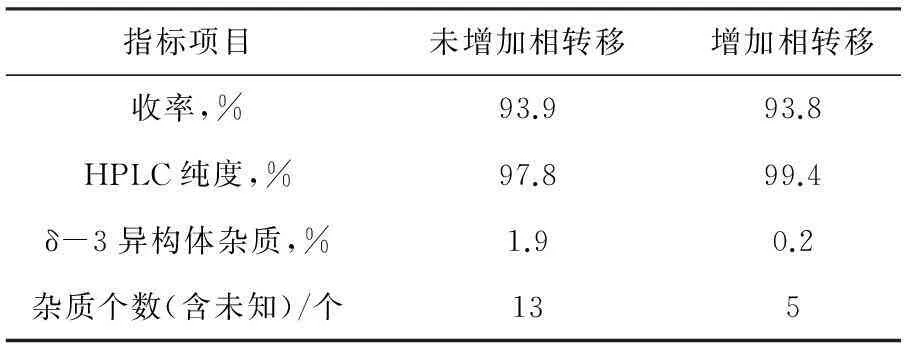
指標項目未增加相轉移增加相轉移收率,%93.993.8HPLC純度,%97.899.4δ-3異構體雜質,%1.90.2雜質個數(含未知)/個135
1.5計算
各個步驟收率計算方法如下,
1) DCC摩爾收率計算方法,以起始投料物7-ACA計:
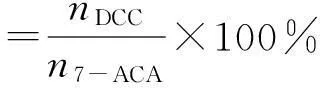
其中YDCC為DCC的摩爾收率,nDCC為DCC的摩爾數,mDCC為DCC實際產出量,MDCC為DCC分子量、n7-ACA為投料物7-ACA的摩爾數,M7-ACA為7-ACA分子量、m7-ACA為7-ACA實際投料量g。
2) 頭孢呋辛酸摩爾收率計算方法,以投料物DCC計:

其中Y頭孢呋辛酸為頭孢呋辛酸的摩爾收率,n頭孢呋辛酸為頭孢呋辛酸的摩爾數,m頭孢呋辛酸為頭孢呋辛酸實際產出量,M頭孢呋辛酸為頭孢呋辛酸分子量。
3) 頭孢呋辛酯摩爾收率計算方法,以投料物頭孢呋辛酸計:

其中Y頭孢呋辛酯為頭孢呋辛酯的摩爾收率,n頭孢呋辛酯為頭孢呋辛酯的摩爾數,m頭孢呋辛酯為頭孢呋辛酯實際產出量,M頭孢呋辛酯為頭孢呋辛酯分子量。
以起始物料7-ACA計,頭孢呋辛酯總的摩爾收率為以上三步收率之積。
2結果與討論
2.1酰氯化試劑三氯氧磷的用量對收率的影響
表2為酰氯化試劑三氯氧磷的用量對DCC收率的影響
表2三氯氧磷用量對反應收率的影響
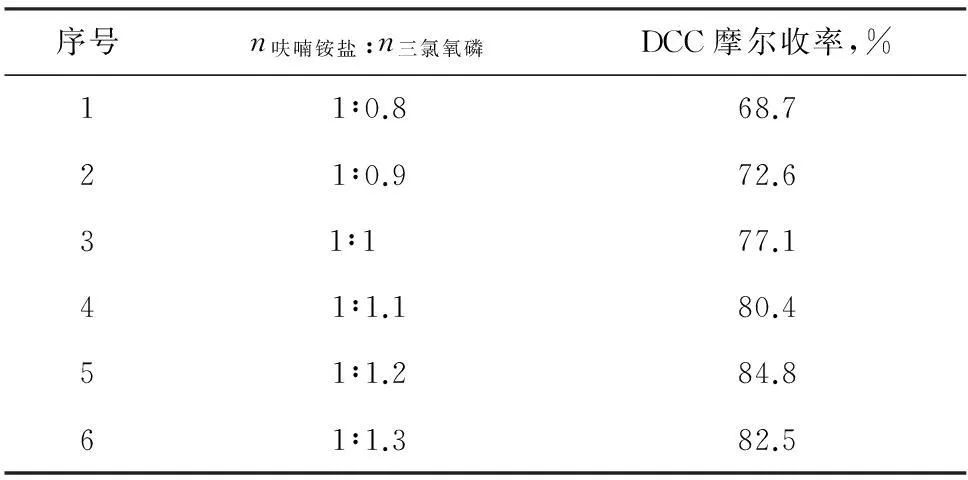
序號n呋喃銨鹽:n三氯氧磷DCC摩爾收率,%11∶0.868.721∶0.972.631∶1 77.141∶1.180.451∶1.284.861∶1.382.5
由表2可知,呋喃銨鹽與三氯氧磷配比為1∶1.2時,最為適宜,當三氯氧磷用量少時反應不完全,收率低;隨著三氯氧磷的增多,收率變化很小,同時由于三氯氧磷投入量太大會造成過量的三氯氧磷對環境造成嚴重危害及生產三廢難處理等問題,故三氯氧磷量優選呋喃銨鹽的1.2倍。
2.2DCC制備過程中鹽酸加料時間對收率的影響
表3為DCC制備過程中鹽酸加料時間對DCC反應收率的影響。
表3鹽酸加料時間對反應收率的影響

序號加料時間/minDCC摩爾收率,%14580.123582.232584.841581.45580.3
由表3可知,加料時間為25 min時,最為適宜。對于鹽酸加料時間縮短時,呋辛酸的收率變高,但當再要縮短時間時,就會適得其反,頭孢呋辛酸的收率會大大降低,這是由于在酸性條件下D-7-ACA的羧基可與其3位的羥基縮合形成內酯(雜質產生反應式見下圖2),導致中間體DCC純度降低,而使最終成品的收率降低。
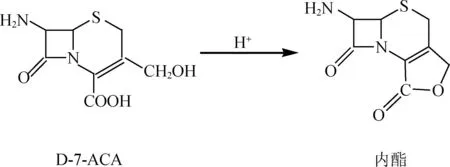
圖2 雜質形成機理
2.3呋辛酸縮合時反應溫度對DCC收率影響
在其他條件不變的情況下,只改變縮合反應溫度,考察溫度對反應收率的影響,具體見表4。
表4頭孢呋辛酸縮合反應溫度對收率的影響

序號縮合溫度/℃反應時間/minDCC摩爾收率,%1-13~-88063.22-18~-138085.13-23~-188089.54-28~-238091.45-33~-288088.2
由表4可見,實驗4較為理想,在此情況下,頭孢呋辛酸的收率較高,實驗1由于反應溫度太高,反應產生雜質太多,最終導致產品收率大幅下降;對于實驗5,由于反應溫度太低,在固定的時間內反應率較低,但在該溫度下,筆者通過延長反應時間進行考察,發現隨著時間的延長,其中相關雜質顯著提高,同時由于溫度的進一步降低,制冷成本將大幅上升,考慮到最終成品質量及成本問題,該反應優選反應溫度-28~-23 ℃。
2.4側鏈1-溴乙基乙酸酯加入后縮合反應溫度對收率影響
在其他條件不變的情況下,只改變側鏈1-溴乙基乙酸酯加入后縮合反應溫度(頭孢呋辛酯縮合反應溫度),觀察對頭孢呋辛酯反應收率的影響,見表5所示。
表5頭孢呋辛酯縮合反應溫度對收率的影響
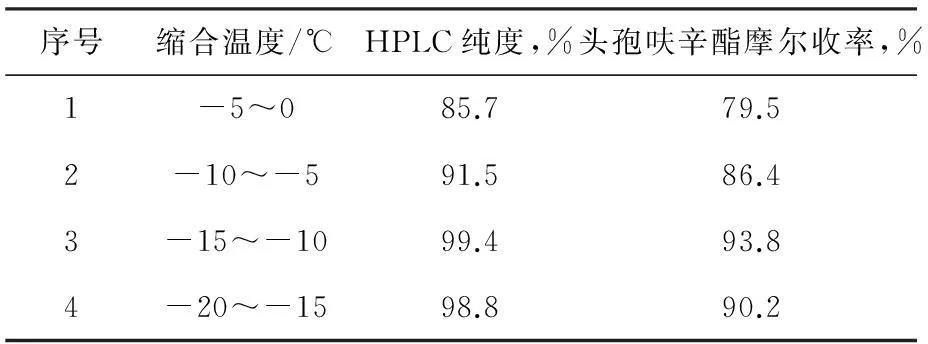
序號縮合溫度/℃HPLC純度,%頭孢呋辛酯摩爾收率,%1-5~085.779.52-10~-591.586.43-15~-1099.493.84-20~-1598.890.2
由表5可見,實驗3較為理想,在此情況下,頭孢呋辛酯的收率較高,實驗1和2由于反應溫度較高,形成大量雜質,一般生成的雜質主要為δ-3異構體(產生機理見下圖3),使得頭孢呋辛酯純度降低的同時收率也大大降低,實驗4反應溫度太低,雖然不會導致反應過程產生大量雜質,但這樣會使得反應不完全,會殘留少量原料,使得純度降低,同時,反應溫度太低,在生產上能耗很大,故考慮到最終的質量、成本以及收率,優選實驗3,即頭孢呋辛酯縮合反應溫度選擇-15~-10 ℃。
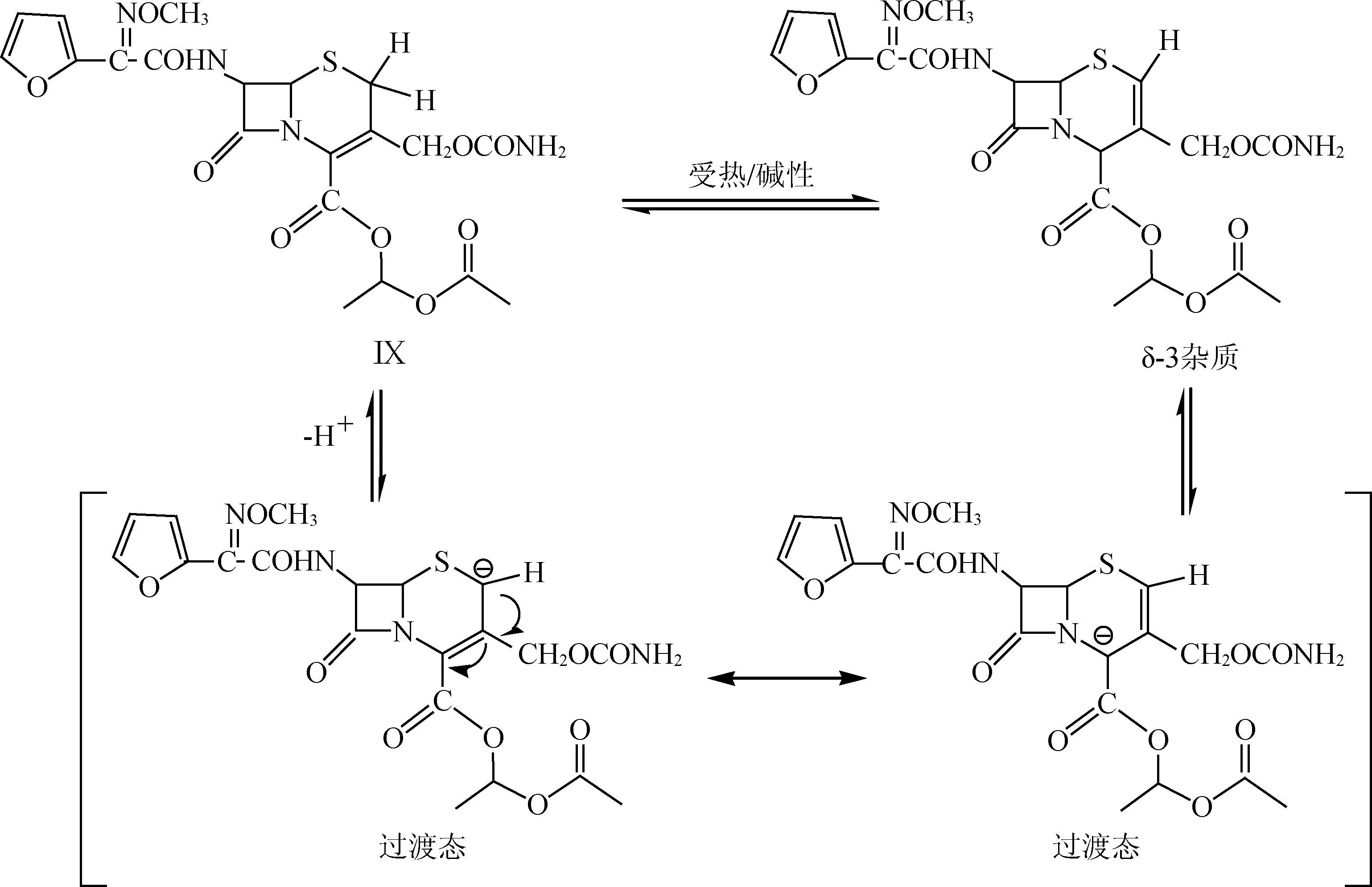
圖3δ-3雜質形成機理
3結論
本研究順利地合成了頭孢呋辛酯,首先,對酰氯化試劑三氯氧磷的用量進行了優化,該過程兼顧降低生產成本和對環境的保護理念,最終得出最優投料量,即nSMIA∶nPOCl3為1∶1.2;其次,對DCC中間體制備過程中鹽酸滴加時間進行優化,該步驟十分關鍵,鹽酸滴加時間在影響質量的同時也會影響到產品收率,經過大量實驗最終得出最優滴加時間25 min;再次,對中間體呋辛酸縮合反應時溫度進行了優化,得最優反應溫度-28~-23 ℃;最后,對頭孢呋辛酯的縮合反應溫度進行優化,確定頭孢呋辛酯縮合反應溫度在-15~-10 ℃。
通過對頭孢呋辛酯合成過程的關鍵點和參數進行了優化,使得中間體Ⅴ、頭孢呋辛酸(Ⅵ)和頭孢呋辛酯成品Ⅸ的收率分別為84.8%、91.4%和93.8%,總收率高達72.7%,該反應具有收率高、成本低、操作容易和環境友好等特點,并具有工業化生產價值。
參考文獻:
[1]鐘宇眉.頭孢呋辛酯片治療尋常痤瘡45例療效觀察[J].中國麻風皮膚病雜志,2011,27(8):592-592.
[2]王久伶,于沛濤,涂銀萍,等.依替米星聯合頭孢呋辛酯治療14~18歲社區獲得性肺炎臨床分析[J].臨床肺科雜志,2013,18(8):1376-1377.
[3]劉躍建,于云芝,李小惠,等.注射用頭孢呋辛酯臨床研究[J].中國抗生素雜志,2002,27(12):734-737.
[4]王榮耕.具有較好市場前景的頭孢類抗生素-頭孢呋辛酯[J].精細化工原料及中間體,2003,(11):15-18.
[5]薛亮.頭孢類抗生素及中間體發展淺析[J].精細化工原料及中間體,2007,(3):27-29.
[6]張軍立,白鵬.頭孢呋辛酯合成方法研究[J].河北工業科技,2006,23(6):328-329.
[7]Bunnel C A.Process for preparing acid halides:US,5084568[P].1992-01-28.
[8]Tsuji T,Okada T.Hydroxymethyl cephem compounds and their preparation:EP,0204517A2[P].1987-09-23.
[9]李愛軍,周雪琴,劉東志.頭孢呋辛鈉合成工藝優化[J].天津大學學報,2007,40(11):1342-1345.
[10] Siviero E,Cabri W,Terrassan D M.Process for the preparation of β-lactam derivatives:US,6458558[P].2002-10-01.
[11] Deshpande P B,Deshpande P N,Khadangale B P,et al.Process for the preparation of cef -uroxime sodium:US,20040092735[P].2004-03-13.
[12] Kremminger P.Intermediates in cephalosporin production:US,20030171577[P].2003-09-11.
[13] Cabri W,Siviero E,Darverlo P L,et al.Process for the synthesis of β-lactam derivatives: US,6642378[P].2003-11-04.
[14] Tyagi O D,Yadav G C,Handa V K.Process for the preparation of highly pure crystalline (R,S)-cefuroxime axetil:US,6833452[P].2004-12-21.
[15] 王建軍,張鵬.一種合成頭孢呋辛酸的方法:中國,104072516A[P].2014-06-18.
[16] 魏青杰,劉東,曹衛凱,等.頭孢呋辛酸、頭孢呋辛酯生產新技術開法[EB/OL].河北省科學技術研究成果公報第四號,河北省科學技術廳,2012,87-88.http://www.hebstd.gov.cn/banshi/tongzhi/kejiting/content_79190.htm
[17] 曹衛凱.頭孢呋辛酸的合成研究[J].化工與醫藥工程,2016,37(2):35-38.
Optimization of synthesis technology of cefuroxime axetil
Cao Weikai
(Xi’anWanlongPharmaceuticalCo.Ltd.,Xi’anShaanxi710119,China)
Abstract:On the synthesis of cefuroxime axetil from 7-aminocephalosporin acid(7-ACA),the synthetic process and critical points were optimized. Firstly,the 7-ACA was hydrolyzed in the low temperature mixture of methanol and water to obtain D-7-ACA,without separation,the acylation reaction was carried out at location C-7 site directly. After crystallization,separation and drying,the crirical intermediate DCC was obtained with the yield of 84.8%. Then through the reaction with chlorosulfonyl isocyanate(CSI),the formamyl transformation at location C-3 had been completed and the solution of cefuroxime acid was obtained. After crystallization,separation and drying,the output yield could reached 91.4%. At last,through the condensation with 1-bromoethyl acetate, the cefuroxime axetil was obtained,with the yield of 93.8%,and the total yield was 72.7%,This process had the characteristics of high yield, low cost, easy to operate, and extremely had the industrial production value.
Key words:cefuroxime acid; cefuroxime axetil; synthesis; industrialization
收稿日期:2016-05-20
作者簡介:曹衛凱(1987-),陜西渭南人,執業藥師,主要從事藥物合成和精細化工領域的研究。
中圖分類號:TQ465
文獻標識碼:A
文章編號:1006-334X(2016)02-0004-05
