GC測定鹽酸帕洛諾司瓊原料藥中(S)-3-氨基-奎寧的含量
2016-08-04 03:48:13曹天海
中國合理用藥探索 2016年3期
曹天海
(昆明積大制藥股份有限公司國際GMP認證部,云南 昆明 650106)
GC測定鹽酸帕洛諾司瓊原料藥中(S)-3-氨基-奎寧的含量
曹天海
(昆明積大制藥股份有限公司國際GMP認證部,云南 昆明 650106)
目的:建立氣相色譜法測定鹽酸帕洛諾司瓊原料藥中(S)-3-氨基-奎寧含量的方法。方法:采用SE-30色譜柱(53 mm×60 m,5 μm),起始溫度100℃,維持5 min,再以20℃/min的速度升溫至180℃,維持15 min,FID檢測器。結果:(S)-3-氨基-奎寧線環性關系良好,相關系數為0.999 7,定量限為0.007 5 mg/mL,檢測限為0.002 5 mg/mL,方法回收率為101.4%,RSD為 3.2%,滿足要求。結論:本法靈敏、可靠,可用于鹽酸帕洛諾司瓊原料藥中(S)-3-氨基-奎寧的質量控制。
鹽酸帕洛諾司瓊;氣相色譜;(S)-3-氨基-奎寧;含量
(S)-3-氨基-奎寧(圖1)是合成鹽酸帕洛諾司瓊的兩個起始物料之一,其合成路線見圖2[1],本品在第2步加入,至最終產品還有2步反應,合成1 g鹽酸帕洛諾司瓊需要加入1.9 g(S)-3-氨基-奎寧。因為(S)-3-氨基-奎寧水溶性較好,不易去除,因此需要考慮它在原料藥中的殘留。由于沒有紫外吸收,一般衍生化后采用高效液相色譜法(HPLC)測定[2]。由于鹽酸帕洛諾司瓊也具有氨基,衍生化時會產生雜質干擾測定。因此,參照《中國藥典》2010年版附錄要求[3],擬定了鹽酸帕洛諾司瓊原料藥中(S)-3-氨基-奎寧的氣相檢測方法,并進行相關的分析方法學驗證,結果表明本方法準確、靈敏、可靠。
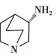
圖1 (S)-3-氨基-奎寧結構式
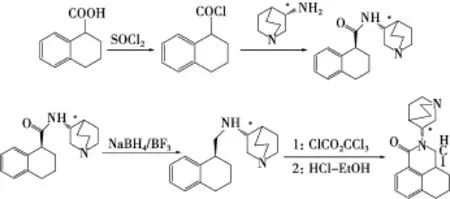
圖2 鹽酸帕洛諾司瓊合成路線
1儀器與試藥
島津 GC-2010氣相色譜儀;FID檢測器;GC-Solution色譜工作站;CTC自動進樣器。
鹽酸帕洛諾司瓊(批號:130901,130902,131001),(S)-3-氨基-奎寧對照品(昆明積大制藥股份有限公司提供)。
2方法與結果
2.1溶液配制
取本品適量,精密稱定,加水制成每1 mL中含50 mg的溶液,作為供試品溶液。
精密稱取(S)-3-氨基-奎寧適量,加水制成每1 mL中含0.05 mg的溶液,搖勻,即得。作為對照品溶液,對照品溶液色譜圖見圖3。
2.2色譜條件
色譜柱:SE-30(53 mm×60 m,5 μm)(蘭州物化所);柱溫:起始溫度100℃,維持5 min,再以20℃/min的速度升溫至180℃,維持15 min;進樣口溫度:240℃;FID檢測器溫度:250℃;載氣:N2;分流比:5∶1;H2流速:38.5 mL/min;Air流速:400 mL/min;尾吹氣:N2,3 mL/min;進樣體積:2.0 μL。理論塔板數按(S)-3-氨基-奎寧計算應不低于5 000。
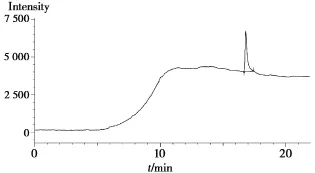
圖3 (S)-3-氨基-奎寧GC色譜圖
2.3專屬性試驗
取濃度為0.05 mg/mL的(S)-3-氨基-奎寧水溶液進樣,主峰保留時間為16.809 min。取純化水進樣,純化水沒有干擾,(S)-3-氨基-奎寧主峰理論板數為66 517,拖尾因子為2.5,符合要求。
2.4精密度試驗
取濃度為0.05 mg/mL的(S)-3-氨基-奎寧水溶液連續進樣6次,計算(S)-3-氨基-奎寧主峰峰面積的RSD值為2.9%,小于5.0%,說明本法重復性好。
2.5檢測限與定量限
取“2.3”中(S)-3-氨基-奎寧溶液0.5 mL,置于10 mL容量瓶中,加水稀釋至刻度,作為檢測限溶液,取2 μL進樣,信噪比約為3∶1,得出(S)-3-氨基-奎寧檢測限為0.002 5 mg/mL;取“2.3”中(S)-3-氨基-奎寧溶液1.5 mL,置于10 mL容量瓶中,加水稀釋至刻度,作為定量限溶液,取2 μL進樣,信噪比約為10∶1,得出(S)-3-氨基-奎寧定量限為0.007 5 mg/mL。結果表明本法靈敏可靠。
2.6線性和范圍
取(S)-3-氨基-奎寧25 mg,置于50 mL量瓶中,加水稀釋至刻度,分別精密移取0.3,0.5,1.0,2.0,3.0 mL,分別置于 10 mL容量瓶中,加水稀釋至刻度。分別取2 μL進樣,計算相關系數為0.999 7。
2.7準確度試驗
取本品已測定(S)-3-氨基-奎寧的原料藥1批,加水制成每1 mL中含有0.522 mg鹽酸帕洛諾司瓊的溶液,分別精密移取1 mL,置10 mL量瓶中,共制備9份,分別加入濃度為0.048 5 mg/mL的(S)-3-氨基-奎寧溶液0.4,0.5,0.6 mL各3份;依法測定,計算回收率,平均回收率為 101.4%,RSD為3.2%。
2.8樣品測定
本品3批樣品(批號:130901,130902,131001)對(S)-3-氨基-奎寧進行了測定,結果均未檢出。
3討論
(S)-3-氨基-奎寧具有一定毒性,易溶于水,為鹽酸帕洛諾司瓊母環的一部分,性質與其相似,必須測定其在本品中的殘留。
由于本品無紫外吸收,本文采用HPLC,先后試用了示差折光檢測器(RID)和蒸發光散射檢測器(ELSD),但均未出現色譜峰,因此只能選用GC方法,無須進行衍生化試驗,測定結果更為準確、快速。
[1]楊新華,歐陽強.鹽酸帕洛諾司瓊合成路線圖解[J].中國醫藥工業雜志,2009,40(3):231-233.
[2] Demian I,Gripshover DF.Enantiomeric purity determination of 3-aminoquinuclidine by diastereomeric derivatization and highperformance liquid chromatographic separation[J].J Chromatography,1989,(466):415-420.
[3] 國家藥典委員會.中華人民共和國藥典(二部)[M].北京:中國醫藥科技出版社,2010:附錄61-65.
Opicapone輔助左旋多巴用于帕金森病和劑末運動波動患者有益
《柳葉刀神經病學》雜志于 2016年2月第 15卷第2期發布了一項關于Opicapone輔助左旋多巴(通用名:Levodopa)用于帕金森病和劑末(endof-dose)運動波動患者的臨床試驗內容。
Opicapone為在研、一日 1次、第三代強效兒茶酚胺氧位甲基轉移酶抑制藥。本項試驗旨在評估Opicapone輔助左旋多巴用于帕金森病和劑末運動波動患者與安慰劑或恩他卡朋相比的安全性和有效性。
本項試驗為隨機、雙盲、安慰劑對照及活性對照試驗。受試者按1∶1∶1∶1∶1的比例隨機分入Opicapone 5 mg組、Opicapone 25 mg組、Opicapone 50 mg組、安慰劑組或恩他卡朋組,用藥14~ 15周。試驗的首要終末指標為基線至治療結束后“關”狀態絕對時間的變化,經患者日記評定。主要分析后,進行分層步驟,首先在全面分析中評定3 種Opicapone劑量與安慰劑相比是否優效,如優效,則在符合方案分析中評定 3種 Opicapone劑量與恩他卡朋相比是否非劣效,非劣效性界限為30分鐘。
試驗結果顯示,安慰劑組“關”狀態時間平均減少56分鐘,恩他卡朋組96.3分鐘,Opicapone 5 mg組91.3分鐘,Opicapone 25 mg組85.9分鐘,Opicapone 50 mg組116.8分鐘。Opicapone 50 mg療效優于安慰劑,不劣于恩他卡朋。Opicapone 5 mg組或25 mg組療效與安慰劑組無顯著差異。最常見的不良事件有運動障礙、失眠、便秘。
總體而言,Opicapone 50 mg輔助左旋多巴用于帕金森病和劑末運動波動患者,可潛在降低左旋多巴日劑量,延長用藥間隔,使用藥獲益最大化。
(來源:http://www.thelancet.com)
Determination of(S)-3-Aminoquinuclidine Content in Drug Substance of Palonosetron Hydrochloride by GC
Cao Tianhai
(International GMP Certification Department,Kunming Jida Pharmaceutical Co.,Ltd.,Yunnan Kunming 650106,China)
Objective:To develop an GC method for determination of(S)-3-aminoquinuclidine content in drug substance of palonosetron hydrochloride.Methods:A SE-30 column(53 mm ×60 m,5 μm)was used at an initial temperature of 100℃ for 5 min,then the temperatureincreased at a rate of 20℃ /min to 180℃ and was maintained for 15 min.The detector used was FID.Results:The calibration curve of(S)-3-aminoquinuclidine showed good linearity,with a correlation coefficient of 0.999 7.The limit of quantitatiion(LOQ)and limit of detection(LOD)of the method were 0.007 5 and 0.002 5 mg/mL respectively,while the recovery rate was 101.4%,with a RSD of 3.2%,which met the relevant requirements.Conclusion:The developed method was sensitive and reliable,which was applicable for the quality control of(S)-3-aminoquinuclidine in drug substance of palonosetron hydrochloride.
Palonosetron Hydrochloride;GC;(S)-3-Aminoquinuclidine;Content
10.3969/j.issn.1672-5433.2016.03.007
曹天海,男,碩士,工程師。主要從事藥物研發工作。E-mail:skyrin_jida@126.com
2015-11-26)
