氯化條件對正己烷異構化Pt/Al2O3催化劑性能的影響
2016-08-12 01:01:28張孔遠王宗波劉晨光
石油學報(石油加工) 2016年4期
張孔遠, 李 勇, 王宗波, 劉晨光
(中國石油大學 重質油國家重點實驗室 CNPC催化重點實驗室, 山東 青島 266580)
?
氯化條件對正己烷異構化Pt/Al2O3催化劑性能的影響
張孔遠, 李勇, 王宗波, 劉晨光
(中國石油大學 重質油國家重點實驗室 CNPC催化重點實驗室, 山東 青島 266580)
摘要:采用浸漬法制備了Pt/Al2O3催化劑,在250和300℃下氯化不同時間,得到兩個系列的Pt/Al2O3-Cl催化劑。采用BET、NH3-TPD、Py-IR等方法表征了制備的催化劑。以正己烷為原料,在反應溫度140℃、壓力2.0 MPa、體積空速1.2 h-1、氫/油摩爾比2的條件下,考察了Pt/Al2O3-Cl催化劑的異構化催化活性和異構產物選擇性。結果表明,在相同溫度下,氯化時間越長,Pt/Al2O3-Cl的比表面積越小,孔徑越大。在250℃氯化時,氯化時間越長,催化劑酸量和氯質量分數(shù)越高,催化正己烷異構化活性和異構產物選擇性越高;但在300℃氯化時,氯化時間超過1 h,所得Pt/Al2O3-Cl的酸量和氯質量分數(shù)下降,催化性能變差。氯化后的Pt/Al2O3-Cl催化劑只有L酸位,經(jīng)異構化反應評價后催化劑既有L酸又有B酸。300℃氯化1 h最佳,所得Pt/Al2O3-Cl催化正己烷異構化的轉化率為84.05%,2,2-二甲基丁烷選擇性為26.54%;連續(xù)運行300 h,催化劑活性幾乎不改變。
關鍵詞:Pt/Al2O3; 四氯化碳; 氯化; 異構化; 酸性
隨著環(huán)境保護的日益嚴格,各國紛紛出臺了新的環(huán)境法規(guī),對汽油質量和汽車尾氣排放標準提出了越來越嚴格的要求,汽油開始向無鉛、低芳烴、低蒸氣壓、低烯烴、低硫、高辛烷值方向發(fā)展,輕質烷烴的異構化技術得到迅速的發(fā)展[1-2]。在工業(yè)上,輕質烷烴異構化催化劑主要以UOP和Axens公司開發(fā)的催化劑為代表。輕質烷烴異構化催化劑可以分成3類。一類是低溫氯化鋁型催化劑,該催化劑以Al2O3為載體,金屬Pt、Pd為活性組分,氯化劑補氯,反應溫度在110~180℃,催化劑的操作溫度低,選擇性好,活性最高,但該催化劑對H2O、S、N等毒物較為敏感,不易再生。具有代表性的催化劑是UOP公司的I-82、I-84催化劑和Axens的IS614A、ATIS-2 L催化劑[3]。第2類是中溫分子篩型催化劑,該催化劑采用分子篩提供酸性組分,金屬Pt、Pd為活性組分,催化劑使用過程中不需要補氯,反應溫度在210~280℃,該催化劑對原料中的毒物耐受程度較高,可以再生。具有代表性的催化劑是UOP公司的HS-10和Axens公司的IP-632催化劑。第3類催化劑是固體超強酸型催化劑,該催化劑是一種新型硫酸化金屬氧化物催化劑,金屬氧化物為SnO2、ZrO2和Fe2O3,加入硫酸或硫酸銨進行酸化,反應溫度比中溫型催化劑低約80℃,活性高,不易失活,可再生。該催化劑主要代表為UOP的PI-242[4]。由于異構化反應是放熱反應,低溫反應更有利于達到反應平衡,因此,低溫異構化技術在工業(yè)上得到廣泛應用。
筆者以Al2O3為載體,金屬Pt為活性組分,CCl4作為氯化劑,正己烷作為異構化反應的模型化合物,采用BET、NH3-TPD、Py-IR等表征手段,研究了不同氯化條件對催化劑催化正己烷異構化反應的轉化率和2,2-二甲基丁烷(2,2-DMB)選擇性的影響,并考察了催化劑活性穩(wěn)定性。
1 實驗部分
1.1原料和試劑
磷酸、氯鉑酸、檸檬酸、濃硝酸、乙醇、濃鹽酸、正己烷、四氯化碳、硝酸銀、丙三醇、硫酸鐵、氰化鉀、過氧化氫溶液、氯化錫,分析純,國藥集團化學試劑公司產品;SB粉,工業(yè)一級,德國Sasol公司產品;N2、H2,體積分數(shù)99.999%,青島恒盛氣體有限公司產品。
1.2催化劑的制備
以SB粉為原料,加入助擠劑與成型劑,擠條成型,經(jīng)干燥、焙燒得到γ-Al2O3載體。以氯鉑酸作為活性組分的前驅物,采用等體積浸漬法浸漬γ-Al2O3,經(jīng)干燥、焙燒制得Pt質量分數(shù)為0.4%的Pt/Al2O3催化劑。所制備的Pt/Al2O3催化劑的物化性質見表1。

表1 Pt/Al2O3的物化性質
1.3催化劑的氯化
將5 mL催化劑置于反應管中,450℃下還原2 h,降溫至250℃,N2作為載氣帶入飽和的CCl4蒸汽,分別氯化1、1.5、2、4和6 h,得到第1系列催化劑;同樣條件還原后降溫至300℃,分別氯化0.5、1、1.5和2 h,得到第2系列催化劑。
1.4催化劑的表征
采用美國麥克公司Micromeritics型自動吸附儀進行低溫N2吸脫附法(BET)表征。樣品在573 K下脫氣抽真空預處理,然后回充N2至常壓,準確稱量樣品質量進行測定。
采用美國Quanta chrome CHEMBET-3000型TPD/TPR分析儀進行NH3-TPD表征。樣品在423 K 下He氛圍脫水0.5 h,降溫至373 K下吸附氨氣至飽和,然后He氣吹掃,直至導熱監(jiān)測(TCD)基線平穩(wěn),然后升溫至700℃,記錄NH3-TPD 曲線。根據(jù)脫峰溫度以及脫峰面積判斷酸的強弱和酸量。
采用美國尼高力Nicolet-58SXC型傅里葉變換紅外光譜儀進行Py-IR表征。樣品需要在150℃下脫水預處理,一部分在真空條件下吸附吡啶,然后在真空干燥箱內150℃物理脫附;另一部分作為空白樣。漫反射紅外掃描波長范圍為4000~400 cm-1。
采用佛爾哈德法(Volhard method) 測定Cl的質量分數(shù)。稱量0.1 g催化劑,使用50 mL的6 mol/L 濃硝酸溶解,加入2 mL質量分數(shù)10%的Fe2(SO4)3指示劑以及5 mL的0.1 mL/L AgNO3,用0.1 mL/L的KSCN進行返滴定。
采用紫外分光光度法測定催化劑Pt質量分數(shù)。取配制好的鹽酸約5 mL、磷酸溶液5 mL,并滴入數(shù)滴H2O2置于100 mL燒杯中,稱取研磨的樣品0.2~0.3 g加入其中,加熱溶解直至溶液呈透明狀態(tài),冷卻后移入50 mL容量瓶中,用去離子水稀釋并搖勻。取上述10 mL溶液、10 mL的鹽酸和10 mL 的質量分數(shù)25%的SnCl2溶液于50 mL的容量瓶中,加入適量的HCl溶液稀釋并搖勻,放置一段時間后在波長460 nm處測定其紫外吸光度,并根據(jù)標準曲線計算溶液的Pt含量,從而推算出催化劑的Pt含量。
1.5催化劑活性評價
在反應壓力2.0 MPa、體積空速1.2 h-1、反應溫度140℃、氫/油摩爾比為2的條件下進行正己烷異構化反應,評價催化劑的異構化活性。分別按式(1)和式(2)計算正己烷轉化率和2,2-二甲基丁烷選擇性。
(1)
(2)
式(1)、式(2)中,x為正己烷轉化率,%;n(C6H14)和n′(C6H14)分別為正己烷進料的物質的量和剩余正己烷的物質的量,mol;s(2,2-DBB)為2,2-二甲基丁烷的選擇性,%;n(2,2-DBB)為反應生成的2,2-二甲基丁烷的物質的量,mol。
2 結果與討論
2.1氯化條件對Pt/Al2O3-Cl催化劑物性的影響
不同溫度、不同時間氯化處理的Pt/Al2O3-Cl催化劑的比表面積以及孔徑列于表2。

表2 不同氯化條件下Pt/Al2O3-Cl催化劑的孔徑和比表面積
從表2看出,在較低的氯化溫度以及較短的氯化時間下,催化劑的比表面積減少較小;隨著氯化時間增加,比表面積下降顯著。氯化溫度對于催化劑的比表面積影響顯著,在300℃下氯化的催化劑的比表面積均小于250℃氯化后的比表面積。在氯化過程中CCl4與Al2O3的表面作用破壞了催化劑的孔結構,尤其是催化劑的小孔遭破壞,或者生成的AlCl3堵塞催化劑的小孔,在很大程度上降低了比表面積[5-6]。李勇等[7]采用FT-IR手段表征了氯化前后的催化劑,發(fā)現(xiàn)在3778、3716及3575 cm-1處紅外吸收峰消失,在3666 cm-1處峰的強度和寬度減弱,表明催化劑表面的羥基受到破壞,催化劑孔結構也會受到破壞。
氯化溫度越高,氯化時間越長,則催化劑的平均孔徑越大。在氯化過程中CCl4與Al2O3的表面作用,破壞了催化劑的孔結構,尤其是催化劑中的小孔,同時還會有大量的AlCl3生成;在測量催化劑孔結構的過程中,催化劑需要在300℃進行脫氣處理,生成的AlCl3會升華,從而增大了孔徑[8]。
2.2氯化條件對Pt/Al2O3-Cl催化劑酸性的影響
2.2.1對酸類型的影響
CCl4與Al2O3之間的化學反應十分復雜。30℃時,Al2O3表面羥基同CCl4就有相互作用;升高溫度時氣相中出現(xiàn)HCl、CO2、CO、COCl2等,γ-Al2O3表面的O以及OH就會被Cl取代,從而改變催化劑的酸性[9-10]。圖1給出了Al2O3載體、氯化后得到的Pt/Al2O3-Cl樣品和活性評價后的樣品的Py-IR譜。

圖1 γ-Al2O3、氯化 Pt/Al2O3-Cl及其活性 評價后樣品的Py-IR譜Fig.1 The Py-IR spectra of γ-Al2O3,chloridized Pt/Al2O3-Cl and its used sample (1) γ-Al2O3; (2) The chloridized Pt/Al2O3-Cl; (3) The used Pt/Al2O3-Cl
從圖1可知,γ-Al2O3只出現(xiàn)了1450 cm-1處吸收峰,表明其上只有L酸;氯化后的Pt/Al2O3-Cl催化劑也在1450 cm-1處出現(xiàn)吸收峰,且峰的強度較γ-Al2O3的大,1540 cm-1處無吸收峰,表明氯化后的Pt/Al2O3-Cl催化劑也只有L酸,無B酸,且L酸量增加,因為Cl取代γ-Al2O3表面的—OH形成Al—O—AlCl2,能夠產生較強的L酸位;催化正己烷異構化反應后的Pt/Al2O3-Cl催化劑在1450以及1540 cm-1處均出現(xiàn)吸收峰,表明其既具有L酸又有B酸。在氫氣存在下,Pt/Al2O3-Cl上部分Cl脫除后生成HCl,與L酸位作用形成B酸位[11-12],具體反應如式(3)、式(4)所示。
(3)
||
(4)
在催化異構化反應的過程中,Pt/Al2O3-Cl會有部分Cl流失,為了維持催化劑的活性和異構產物的選擇性,工業(yè)上需要在進料中補入少量的CCl4。
2.2.2對酸量的影響
由于Cl取代催化劑表面羥基,Al離子與2種類型的陰離子相連。Cl的出現(xiàn)打破了原有的電子對稱性,由于Cl的誘導效應,吸引鄰近羥基的電子,使H原子的正電性增加,在一定程度上提高了催化劑的酸量[13]。圖2為250和300℃不同氯化時間的Pt/Al2O3催化劑的NH3-TPD曲線。

圖2 250℃和300℃下不同氯化時間的Pt/Al2O3-Cl催化劑的NH3-TPD曲線Fig.2 The NH3-TPD profiles of Pt/Al2O3-Cl chloridized at 250℃ and 300℃ for different chlorination time (a) 250℃;(b) 300℃
由圖2可知,300℃氯化后的催化劑在220℃、340℃以及400℃附近出現(xiàn)NH3的脫附峰,其中以220℃歸屬于中等酸的脫附峰為主,在340℃出現(xiàn)歸屬于中等強酸的脫附肩峰,并在400℃出現(xiàn)少量的歸屬于強酸的脫附峰。盡管有微量的弱酸出現(xiàn),氯化后的催化劑還是主要以中強酸為主。
對NH3-TPD出現(xiàn)的NH3脫附峰進行積分,可以半定量比較催化劑的酸量。圖2(a)顯示,250℃氯化的催化劑NH3脫附峰積分面積與氯化時間成正比,即氯化時間越長,積分面積越大,氯化6 h時催化劑酸量最多,氯化1 h時酸量最少,說明在250℃下隨著氯化時間的增長,Al2O3表面的羥基被Cl取代的越多,酸量越多。圖2(b)顯示,與250℃氯化相比,300℃時氯化的催化劑的酸量以及酸強度均有增加。但NH3脫附峰積分面積與氯化時間不成正比,氯化1 h時積分面積最大,1.5 h時次之,2 h 時再次之,0.5 h時最小。這是因為300℃下較短時間內,Al2O3載體表面的羥基會被Cl取代,氯化時間越長羥基取代越多,催化劑的酸性增加,但隨著氯化時間的增加,反應生成部分AlCl3,AlCl3極易升華,造成了Cl的損失,酸量也就隨之降低。
2.3氯化條件對Pt/Al2O3-Cl催化劑氯質量分數(shù)的影響

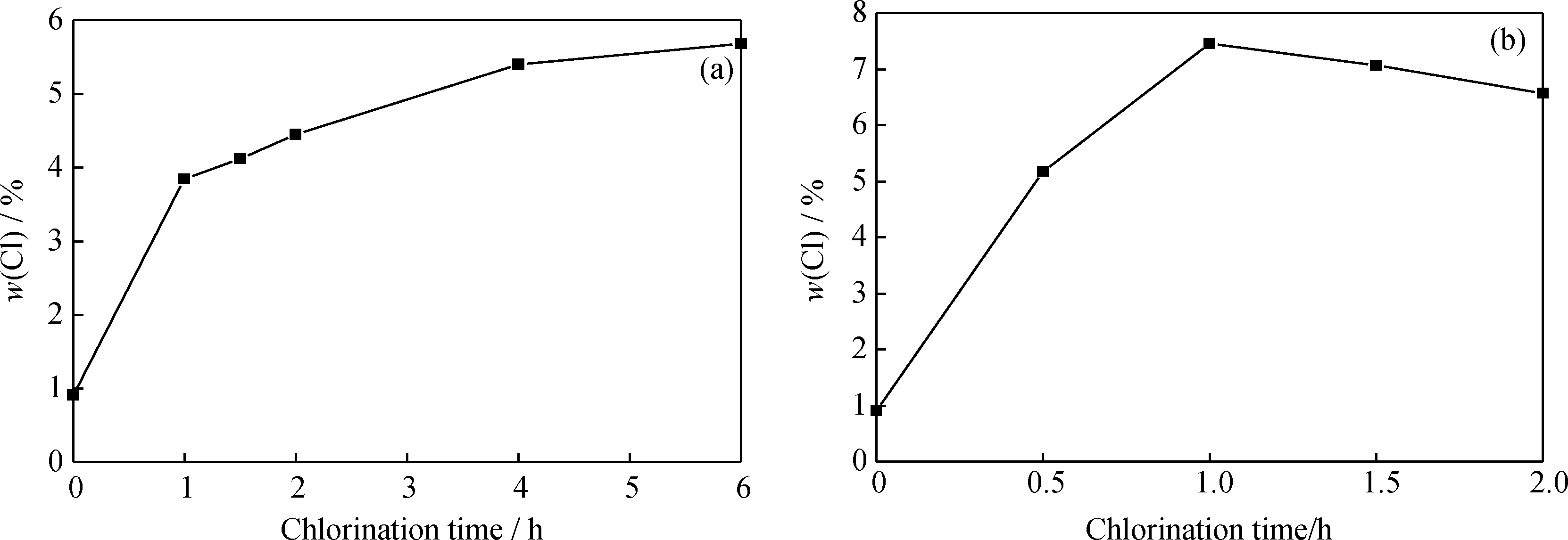
圖3 250℃和300℃下氯化時間對Pt/Al2O3-Cl催化劑Cl質量分數(shù)的影響Fig.3 Effect of chlorination time on Cl mass fraction of Pt/Al2O3-Cl chloridized at 250℃ and 300℃ (a) 250℃; (b) 300℃
2.4氯化條件對Pt/Al2O3-Cl催化劑異構化催化性能的影響
2.4.1對異構化催化活性和異構產物選擇性的影響
以不同氯化條件制備的Pt/Al2O3-Cl為催化劑,在反應壓力2.0 MPa、體積空速1.2 h-1、反應溫度140℃、氫/油摩爾比2條件下催化正己烷異構化反應,正己烷轉化率和2,2-二甲基丁烷選擇性結果列于表3。采用紫外分光光度計測量吸光度[16-17],計算得到的250和300℃下氯化不同時間后催化劑的Pt質量分數(shù)也列于表3。

表3 不同氯化條件所得Pt/Al2O3-Cl催化正己烷異構化反應的轉化率和2,2-二甲基丁烷選擇性及其Pt質量分數(shù)
T=140℃;p=2.0 MPa; LHSV=1.2 h-1;n(H2)/n(Oil)=2
由表3可知,250℃氯化的Pt/Al2O3-Cl催化正己烷異構化的轉化率和2,2-二甲基丁烷選擇性均隨著氯化時間的增加而增加,但當氯化溫度為300℃時,所得轉化率與選擇性先增加后減小,與催化劑的Cl質量分數(shù)變化趨勢幾乎相同。300℃下氯化的催化劑催化正己烷異構化的評價結果優(yōu)于250℃下的,在300℃氯化1 h的Pt/Al2O3-Cl催化劑為最佳。
當氯化溫度為250℃時,氯化過程主要為Cl取代Al2O3的表面羥基,氯化時間越長,催化劑Cl質量分數(shù)越高,催化劑的酸性也越高,正己烷的轉化率與選擇性也越高。在300℃下,CCl4會取代Al2O3表面更多的羥基,酸量較多,酸性較強,但氯化時間的進一步增加會生成極易升華的AlCl3,導致Cl質量分數(shù)的降低,酸量有所減小;另外,AlCl3還會與Pt的氯化物生成PtCl2·2AlCl3,該物質易揮發(fā),遷移和集聚,使得金屬Pt質量分數(shù)降低,分散度下降,在異構化過程中金屬功能減弱,從而降低了轉化率與選擇性,與此同時還會促使催化劑因積炭結焦而失活[18-19]。
在催化劑的氯化過程中,Cl不僅與催化劑的載體發(fā)生反應,而且會與催化劑上的貴金屬反應。由表3可見,在250℃不同氯化時間下,催化劑上的Pt質量分數(shù)幾乎不發(fā)生改變,維持在0.4%左右。但在300℃氯化的催化劑上Pt的質量分數(shù)隨氯化時間的延長呈現(xiàn)下降的趨勢。在使用CCl4氯化處理Pt/Al2O3催化劑時,產生的COCl2會吸附在金屬Pt上,最后形成PtCl2,金屬Pt以+2價態(tài)的形式出現(xiàn)[20]。在250℃下氯化催化劑,CCl4與γ-Al2O3反應, Cl取代催化劑表面的羥基,不影響催化劑的Pt質量分數(shù);隨著反應溫度的升高,CCl4與γ-Al2O3反應生成AlCl3,AlCl3與PtCl2反應生成PtCl2·2AlCl3,該物質易升華,造成金屬Pt與Cl都損失。
2.4.2催化劑的活性穩(wěn)定性
將300℃氯化1 h的Pt/Al2O3-Cl催化劑在微反裝置中連續(xù)反應300 h,考察正己烷的轉化率以及2,2-二甲基丁烷選擇性隨時間的變化,結果示于圖4。
由圖4可知,催化劑在連續(xù)運行300 h過程中,催化劑的活性幾乎沒有發(fā)生變化,正己烷的轉化率和 2,2-二甲基丁烷的選擇性分別維持在83%和25%左右,表明催化劑具有良好的穩(wěn)定性。
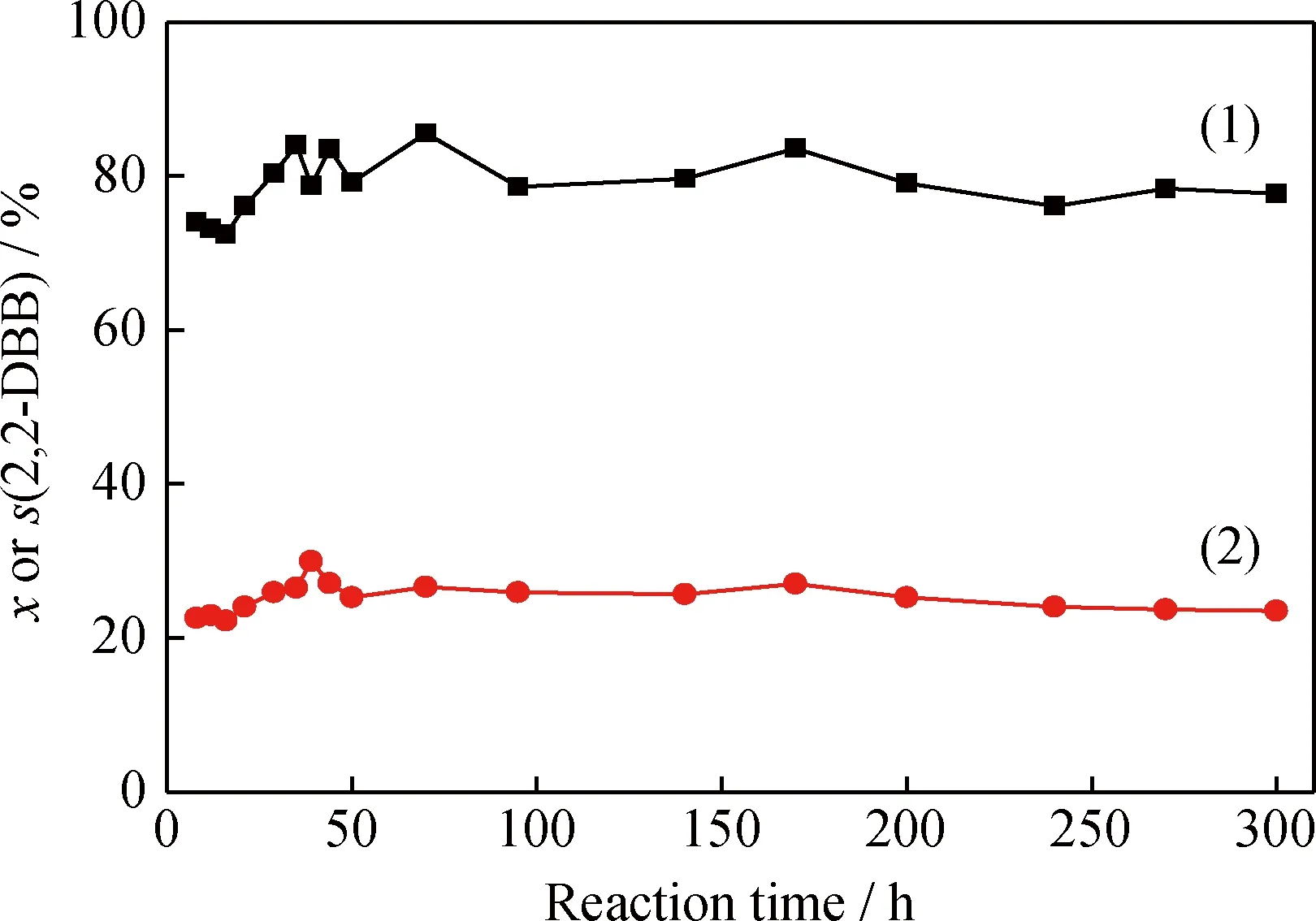
圖4 Pt/Al2O3-Cl催化正己烷異構化反應300 h的 轉化率和2,2-二甲基丁烷的選擇性Fig.4 The conversion and 2,2-DBB selectivity ofn-hexane isomerization over the Pt/Al2O3-Cl catalyst during 300 h continuous operationT=140℃; p=2.0 MPa; LHSV=1.2 h-1; n(H2)/n(Oil)=2 Catalyst chlorination conditions: Temperature of 300℃; Time of 1 h (1) x; (2) s(2,2-DBB)
3 結 論
(1)同一溫度下進行氯化,氯化時間越長,所得Pt/Al2O3-Cl催化劑的比表面積越小,孔徑越大;300℃下氯化的影響比250℃下的顯著。
(2)氯化后的Pt/Al2O3-Cl催化劑上只有L酸位,同Al2O3載體的酸類型一樣,但反應過后的催化劑上既有L酸又有B酸,表明烷烴是在催化劑B酸位進行。
(3)經(jīng)氯化后的Pt/Al2O3-Cl催化劑的酸強度增加,并以中強酸為主。在250℃下,氯化時間越長,則催化劑的酸量越高,Cl質量分數(shù)越高,催化正己烷異構化的轉化率與2,2-二甲基丁烷選擇性越高。但在300℃下,氯化時間超過1 h后,Pt/Al2O3-Cl催化劑的酸量以及Cl質量分數(shù)開始下降,催化劑活性降低。
(4)氯化條件以300℃氯化1 h最佳,Pt/Al2O3-Cl催化劑的酸量以及酸強度最高,Cl質量分數(shù)為7.45%,催化正己烷異構化的轉化率為84.05%,2,2-二甲基丁烷選擇性為26.54%,且具有良好的穩(wěn)定性, 300 h內催化劑活性不發(fā)生改變。
參考文獻
[1] 程偉,程曉晶,王雪楓.直餾汽油異構催化劑的研究進展[J].化工進展,2013, 32(2): 333-339. (CHENG Wei, CHENG Xiaojing, WANG Xuefeng. Advance in straight run gasoline isomerization catalysts[J].Chemical Industry and Engineering Progress, 2013, 32(2): 333-339.)
[2] 陶海橋,宋清虎.多產異構烷烴的工藝研究進展[J].精細石油化工進展, 2005, 6(4): 54-58. (TAO Haiqiao, SONG Qinghu. Study on new process for maximizing isoparaffin[J].Advances in Fine Petrochemicals, 2005, 6(4): 54-58.)
[3] 孫懷宇,陳集,賈增江,等. C5/C6烷烴異構化機理與催化劑研究進展[J].化工時刊, 2005, 19(1): 48-50. (SUN Huaiyu, CHEN Ji, JIA Zengjiang, et al. Research development in C5/C6paraffin isomerization mechanism and its catalyst[J].Chemical Industry Times, 2005, 19(1): 48-50.)
[4] 徐鐵鋼,賈鵬飛,董春明,等. C5/C6正構烷烴異構化催化劑研究進展[J].化工中間體, 2014, (4): 16-19.(XU Tiegang, JIA Pengfei, DONG Chunming, et al. C5/C6paraffin isomerization catalyst and its development[J].Chemical Intermediates, 2014, (4): 16-19.)
[5] GIANNETTI J P,SEBULSKY R T.Chlorinated platinum-alumina low temperature isomerization catalysts[J].Industrial & Engineering Chemistry Product Research and Development, 1969, 8(4): 356-361.
[6] MELCHOR A,GARBOWSKI E, MATHIEU M V, et al. Chlorinated alumina.Acidic properties and catalytic activity towardsn-butane isomerization[J].Journal of the Chemical Society, Faraday Transactions 1: Physical Chemistry in Condensed Phases, 1986, 82(6): 1893-1901.
[7] 李勇,張孔遠,朱紅偉,等. C5/C6烷烴異構化 Pt/Al2O3-Cl 催化劑的性能[J].化工進展, 2015, 34(1): 150-155. (LI Yong, ZHANG Kongyuan, ZHU Hongwei, et al. Performance of Pt/Al2O3-Cl catalysts for C5/C6paraffin isomerization[J]. Chemical Industry and Engineering Progress, 2015, 34(1): 150-155.
[8] JAIN A V,PRADHAN N C,DALAI A K, et al. Studies on chlorided Pt/Al2O3catalysts: Preparation,characterization andn-butane isomerization activity[J].Catalysis Letters, 2003, 86(4): 221-227.
[9] 王囡囡.三氯化鋁固載化催化劑的制備及其催化性能研究[D].大連:大連理工大學, 2006.
[10] 蔡天錫.固載化AlCl3催化劑的研制與應用[J].石油化工, 2001, 30(4): 315-318.(CAI Tianxi. The development and use of immobilized AlCl3catalysts[J].Petrochemical Technology, 2001, 30(4): 315-318.)
[11] DUCOURTY B,SZABO G,DATH J P,et al.Pt/Al2O3-Cl catalysts derived from ethylaluminumdichloride: Activity and stability in hydroisomerization of C6alkanes[J].Applied Catalysis A: General, 2004, 269(1): 203-214.
[12] BERNARD P M, PRIMET M.Influence of hydrogen chloride addition on the catalytic isomerization activity of chlorinated alumina and chlorinated platinum-alumina solids.Superacid behavior[J].J Chem Soc,Faraday Trans, 1990, 86(3): 567-570.
[13] 徐承恩.催化重整工藝與工程[M].北京:中國石化出版社, 2006: 297-300.
[14] CAI T, LIU S,Qü J,et al.Chlorinated alumina and its catalytic behavior in selective polymerization of isobutene[J].Applied Catalysis A: General, 1993, 97(2): 113-122.
[15] CLET G, GOUPIL J M,SZABO G,et al.Chlorinated alumina as an alkylation catalyst: Influence of superficial HCl[J].Journal of Molecular Catalysis A: Chemical, 1999, 148(1): 253-264.
[16] 翁漪.負載型Pt/Al2O3催化劑中鉑的測定[J].工業(yè)催化, 2004, 12(3): 50-52. (WENG Yi. Determination of platinum in Pt/Al2O3catalysts[J].Industrial Catalysis, 2004, 12(3): 50-52.)
[17] 曹蘇薇.國產NDC-4 型脫氫催化劑中鉑含量分析方法的改進[J].化學工程師, 2004,(9): 32-34.(CAO Suwei.The improvement of platinum analysis method for NDC-4 dehydrogenation catalyst[J].Chemical Engineer, 2004,(9): 32-34.)
[18] DELANNAY F,DEFOSSE C,DELMON B, et al.Chloriding of Pt-Al2O3catalysts. Studies by transmission electron microscopy and X-ray photoelectron spectroscopy[J].Industrial & Engineering Chemistry Product Research and Development, 1980, 19(4): 537-541.
[19] MELCHOR A,GARBOWSKI E,MATHIEU M V,et al.Physicochemical properties and isomerization activity of chlorinated Pt/Al2O3catalysts[J].Journal of the Chemical Society, Faraday Transactions 1: Physical Chemistry in Condensed Phases, 1986, 82(12): 3667-3679.
[20] PRIMET M,BASSET J M,MATHIEU M V,et al.Infrared study of CO adsorbed on Pt-Al2O3. A method for determining metal-adsorbate interactions[J].Journal of Catalysis, 1973, 29(2): 213-223.
收稿日期:2015-07-15
基金項目:國家重點基礎研究發(fā)展計劃“973”項目(2010CB226905)和國家自然科學基金項目(21176258和U1162203)資助
文章編號:1001-8719(2016)04-0674-07
中圖分類號:TE624.4
文獻標識碼:A
doi:10.3969/j.issn.1001-8719.2016.04.003
Effect of Chlorination Treatment on Performance of Pt/Al2O3Catalysts forn-Hexane Isomerization
ZHANG Kongyuan, LI Yong, WANG Zongbo, LIU Chenguang
(KeyLaboratoryofCatalysis,CNPC,StateKeyLaboratoryofHeavyOilProcessing,ChinaUniversityofPetroleum,Qingdao266580,China)
Abstract:Pt/Al2O3 catalysts were prepared by impregnation. Two series of Pt/Al2O3-Cl catalysts were obtained from Pt/Al2O3 chloridized at 250℃ and 300℃ for different time, and characterized by BET, NH3-TPD, Py-IR techniques. The catalytic activity of Pt/Al2O3-Cl was evaluated by n-hexane isomerization under a space velocity of 1.2 h-1, 140℃, 2.0 MPa and hydrogen and hexane molar ratio of 2. The results revealed that at the same temperature the longer the chlorination time, the smaller the specific surface area and the larger the pore size of Pt/Al2O3-Cl. The Cl mass fraction, isomerization activity and acid amount of Pt/Al2O3-Cl increased with the increase of chlorination time at 250℃, but decreased as chloridized at 300℃ for more than 1 h. Only Lewis acid sites were detected on fresh Pt/Al2O3-Cl, however, both Lewis and Br?nsted acid sites were detected on used one. The catalytic performance of Pt/Al2O3-Cl chloridized at 300℃ for 1 h was the best for n-hexane isomerization, with the conversion and 2,2-DMB selectivity were 84.05% and 26.54%, respectively, and good stability for 300 h continuous operation without any decline.
Key words:Pt/Al2O3; tetrachloromethane; chloridized; isomerization; acidity
通訊聯(lián)系人: 張孔遠,男,教授級高級工程師,博士,從事石油與天然氣加工專業(yè);Tel:0532-81697776;E-mail:zkyuana@126.com
