煤焦油加氫脫氧精制研究進展
2016-09-02 06:07:32許人軍胡薇月崔文崗李穩宏
廣州化工 2016年15期
許人軍,胡薇月,崔文崗,李 冬,李穩宏
(1 西安市輕工業研究所,陜西 西安 710001;2 陜西延長石油油田化學科技有限責任公司,陜西 延安 716000;3 陜西省產品質量監督檢驗研究院,陜西 西安 710048;4 西北大學化工學院,陜西 西安 710069)
?
煤焦油加氫脫氧精制研究進展
許人軍1,2,胡薇月3,崔文崗4,李冬4,李穩宏4
(1 西安市輕工業研究所,陜西西安710001;2 陜西延長石油油田化學科技有限責任公司,陜西延安716000;3 陜西省產品質量監督檢驗研究院,陜西西安710048;4 西北大學化工學院,陜西西安710069)
簡要介紹了煤焦油中主要含氧化合物的類型,包括酚類、萘類、酯類、酸類、呋喃類和醛類等含氧化合物,綜述了不同類型含氧化合物在加氫脫氧(HDO)過程中的反應機理以及反應路徑,總結了加氫脫氧過程中直接脫氧和間接脫氧的相互關系以及路徑選擇,最后針對煤焦油中含氧化合物復雜及多樣性的特點提出了一些新的改進和研究方向,并對含氧化合物加氫脫氧的未來作了展望。
加氫脫氧;含氧化合物;反應機理;動力學
隨著世界石油資源的匿乏以及能源需求的不斷增加,面對我國“富煤,少氣,缺油”的能源現狀,近年來,人們已經開始尋求和開發新的可替代能源。煤液化油、煤焦油等因其產量不斷增加,越來越引起人們的注意,其可謂理想的能源替代品,然而這些新型可替代能源已存在著諸多加工問題,如高的雜原子含量,相對于原油來說,煤焦油中氧含量極高,這使得在生產清潔燃料油品之前必須將氧脫除[1-4]。因此,對煤焦油進行加氫脫氧研究具有重要的經濟和戰略意義。
本文對主要油品中含氧化合物類型、加氫脫氧反應機理以及加氫脫氧反應動力學的研究進展進行了綜述,并對未來的研究方向進行了展望。
1 含氧化合物的類型
原料中氧含量和氧化物的類型決定了實現較高的加氫脫氧(HDO)轉化率時的氫耗和操作難度[5]。在輕餾分加氫中,HDO并不是很重要,但在重質油加氫催化改質過程中很重要。
HDO是煤液化油生產燃料產品中最重要的反應之一。液化方法和煤的結構決定了氧化物的類型。為了研究其加氫過程中的HDO反應,Gates等[6-8]對由溶劑精煉煤法(SRC)生成的煤液化油進行了大量表征。這些學者使用制備液相層析法從SRC液體里分出了九個餾分段,5,6,7,8-四氫化-1-萘酚,2-羥苯基苯,4-環己基苯基苯酚等酚類化合物主要集中在弱酸餾分段中。其他的含氧化合物,如呋喃類,醚類和酮類集中在中性油餾分段中,在堿性餾分段發現了羥基吡啶和羥基吲哚。
耿層層等[9]對低溫煤焦油中的含氧化合物進行了分析鑒定。此外,吳婷等[10]采用GC-MS及元素分析儀對低溫煤焦油中酸性組分和堿性組分的化學組成和結構進行定性定量分析。其中,酸性組分質量分數不小于0.1%以上的化合物有74種,且全部為含氧化合物,其質量分數為95.4%。
2 加氫脫氧的反應機理
2.1呋喃類
Furimsky[11-13]使用Co-Mo/Al2O3催化劑對呋喃的HDO反應進行了研究,提出了呋喃HDO反應機理,如圖1所示。在上述反應機理中,可利用的表面活化氫是影響反應途徑的決定性因素。途徑1為在一定的反應條件下氧從環中脫出;途徑2為在活化氫濃度更高的情況下,在發生開環反應之前呋喃環產生了部分加氫反應,在后續的反應中生成丙烯或生成丁烯;途徑3為在較高氫分壓下的反應,在這種條件下催化劑表面大部分被活化氫所覆蓋,呋喃環可以完全加氫,主要反應產物丁烷和H2O。
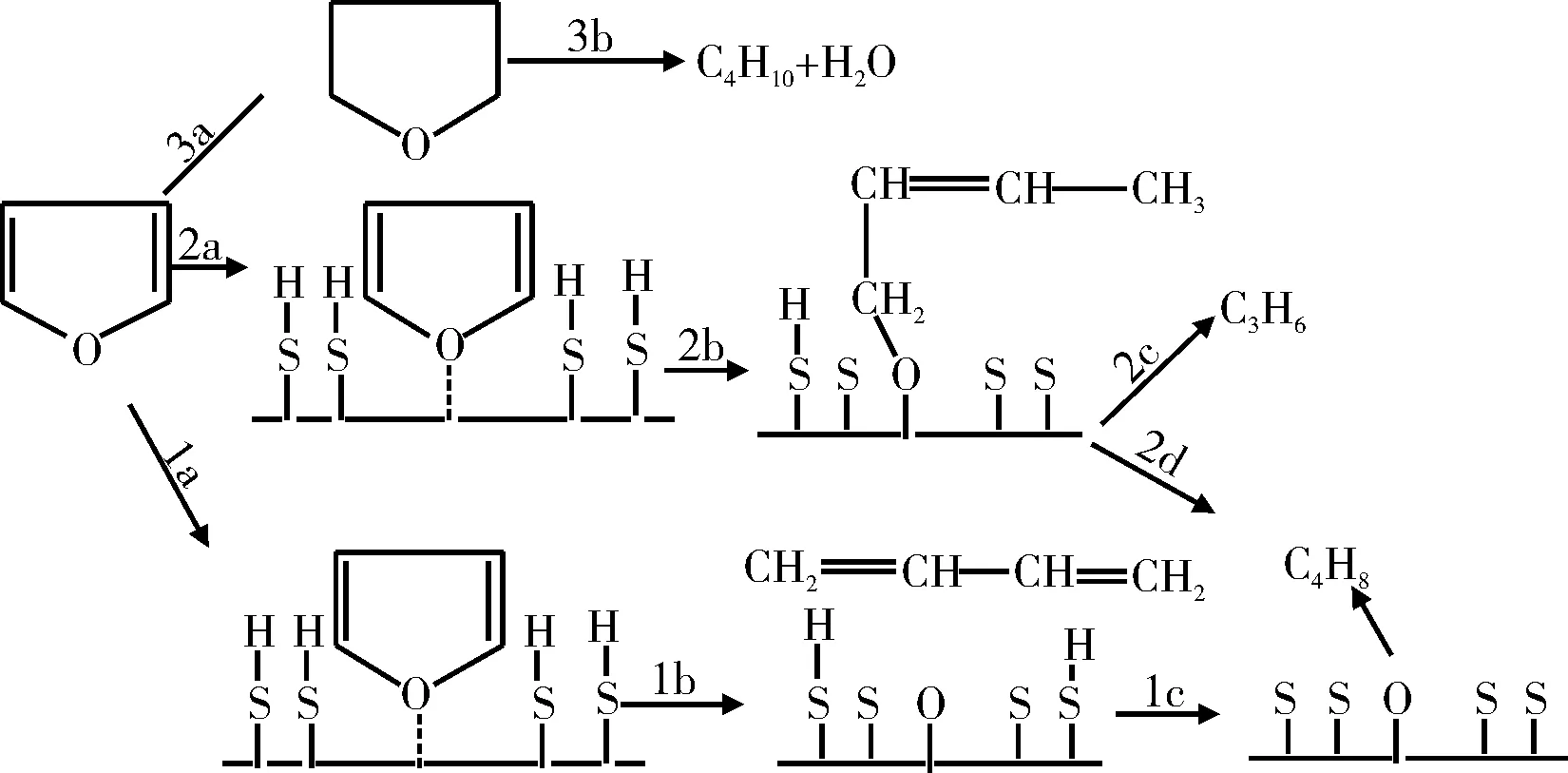
圖1 呋喃HDO反應途徑Fig.1 HDO reaction pathway of furan
Bunch等[14-15]使用硫化態和還原態Ni-Mo/Al2O3催化劑研究了苯并呋喃的HDO反應,提出的反應機理示于圖2。研究結果表明,使用硫化態Ni-Mo/Al2O3催化劑,生成約50%的乙基苯酚及少量的乙苯和乙基環己烷,并且只發現一種加氫含氧中間產物2,3-二氫苯并呋喃。然而,使用還原態Ni-Mo/Al2O3催化劑,苯并呋喃加氫脫氧反應只有氫化反應,經HDO反應生成含氧中間產物的類型較多(如六氫苯并呋喃、2-乙基環己醇和八氫苯并呋喃),中間產物再脫氧生成乙基環己烯和乙基環己烷,產物中并沒有發現乙基苯酚和乙苯。

圖2 苯并呋喃的HDO反應網絡Fig.2 HDO reaction network of benzofuran
Krishnamurthy等[16]根據二苯并呋喃HDO的產物分布和某些中間體的反應活性,提出了二苯并呋喃的HDO反應機理,見圖3。二苯并呋喃的HDO主要按照3種途徑進行:(1)加氫途徑:首先芳環加氫飽和生成含氧中間產物,然后C-O鍵斷裂生成單環烴;(2)氫解途徑:先發生C-O鍵斷裂生成苯基苯酚,再加氫生成單環產物;(3)直接脫氧途徑:二苯并呋喃直接脫氧生成聯苯。在較高的氫分壓下,在雜環開環以前,首先進行的是一個芳環的加氫反應。在反應中生成了鄰-環乙基苯酚中間產物,其進一步脫氧生成二環己烷或是苯基環己烷。這就說明,與相應的含硫化合物HDS反應相比,HDO反應需要更高的氫耗[17]。

圖3 二苯并呋喃HDO反應網絡Fig.3 HDO reaction network of diphenylene-oxide
Satterfield等[18]對苯并呋喃的HDO進行了研究,發現反應條件對產物分布的影響非常顯著。Lavopa等[19]研究了硫化態和氧化態Ni-Mo/Al2O3催化劑對苯并呋喃的加氫脫氧反應。使用硫化態催化劑催化反應,產物中單環烴類占75%,環己烷占主要部分,還有一些重要但含量少的物質如甲基環戊烷、環戊烷、苯、甲基環己烷和環己烯等。然而,使用氧化態催化劑催化反應,單環化合物收率為25%。
2.2酚類化合物
Odebunmi等[20-22]在連續微型反應器內,使用硫化態Co-Mo/Al2O3催化劑在氫分壓為3.0~12.0 MPa下,研究了甲基苯酚的HDO反應途徑,研究發現主要加氫產物是甲苯和環己烷,同時生成了少量環己烯。Samchenko等[23]使用Ni/Cr催化劑研究發現鄰甲基苯酚和對甲基苯酚比間甲基苯酚更穩定。Shin等[24]使用Ni/SiO2催化劑建立了下面的加氫反應順序:苯酚≈間甲基苯酚>對甲基苯酚>鄰甲基苯酚。研究結果均表明,鄰位取代酚的位阻效應影響很大[25]。
Wandas等[26]使用Co-Mo/Al2O3催化劑,在氫氣壓力為7 MPa,溫度為360 ℃條件下研究了甲酚的HDO反應。同樣,直接脫氧生成了許多種類的化合物,且甲基環己烷、甲苯和乙基環戊烷是主要的加氫脫氧產物。Furimsky等[27]采用硫化態Co-Mo/Al2O3催化劑,對鄰位和對位取代苯酚的HDO反應進行了研究。研究結果表明,鄰位取代苯酚的HDO反應活性最弱,苯酚、對乙基苯酚和對叔丁基苯酚HDO反應的轉化率基本相同。鄰位取代苯酚的HDO反應主要按兩種途徑進行如圖4和圖5所示。

圖4 鄰位取代苯酚的HDO反應網絡Fig.4 HDO reaction network of ortho substituted phenol

圖5 對甲基苯酚的HDO反應網絡Fig.5 HDO reaction network of p-methylphenol
Vogelzang等[28]提出的萘酚的HDO反應網絡(圖6),反應網絡中四氫萘酮生成速率最大,表明該反應的最終產物大部分由四氫萘酮轉化而來。該試驗使用硫化態Ni-Mo/Al2O3作為催化劑,反應溫度為200 ℃,氫氣壓力為3.5 MPa。在此條件下,芳香環的加氫飽和比萘酚的直接HDO更容易進行。因此,四氫化萘和5,6,7,8-四氫-1-萘酚占萘酚HDO產物很大一部分比例。然而,在高溫下條件下萘酚的直接HDO速率超過了芳環的加氫飽和。同時,他提出包含1,2-二氫萘酚和四氫萘酮、酮-烯醇的轉化是反應網絡的一部分。而且,順式和反式十氫化萘的生成速率非常低。

圖6 萘酚的HDO反應網絡Fig.6 HDO reaction network of naphthol
徐春華[29]研究了不同反應溫度和壓力下鄰甲酚在硫化態Co-Mo/Al2O3催化劑上的HDO性能,反應中直接脫氧產物甲苯的選擇性高達90%。Lin等[30-31]研究了Rh基催化劑上鄰甲氧基苯酚的HDO反應過程。研究結果表明,鄰甲氧基苯酚在Rh基催化劑上是按先加氫后脫氧的途徑進行反應,反應過程見圖7。王威燕等[32]研究了270 ℃下非晶態催化劑La-Ni-Mo-B催化4-甲基苯酚的HDO反應(見圖8)。在整個反應過程中,沒有檢測到甲苯的生成,這說明在La-Ni-Mo-B催化下,4-甲基苯酚沒有發生直接脫氧反應,而是先加氫生成環己醇,再脫氧生成甲基環己烷,即按照氫化-氫解的途徑進行反應。

圖7 鄰甲氧基苯酚在Rh基催化劑上的HDO反應途徑Fig.7 HDO reaction pathway on Rh based catalysts of o-methoxyphenol

圖8 非晶態催化劑La-Ni-Mo-B催化4-甲基苯酚的HDO反應路徑Fig.8 Reaction scheme of HDO of 4-methyl phenol on La-Ni-Mo-B amorphous catalysts
Bredenberg等[33]在氫壓5 MPa和溫度(275~325)℃條件下,研究硫化態Co-Mo/γ-Al2O3催化劑催化3種甲氧基苯酚異構體的加氫脫氧反應,3種異構體的反應活性順序為對甲氧基苯酚>鄰甲氧基苯酚>間甲氧基苯酚。Kallury等[34]研究了Mo-Ni/Al2O3催化劑催化二羥基苯異構體的加氫脫氧反應,結果表明,間苯二酚活性較弱,鄰苯二酚和對苯二酚的活性比苯酚強,催化脫除一個羥基得到苯酚的收率為60%。
2.3醚類化合物
醚在原料中的含量相對較少,此外,在含羥基的加氫脫氧反應中也可能生成醚。Artok等[35]在溫度范圍為375~425 ℃,氫氣壓力為6.9 MPa,含有MoS2的條件下研究了二苯醚的HDO過程。二苯醚的醚鍵首先加氫裂化生成了苯和酚,并通過將酚轉化苯和環己烷以及環己烷的異構化反應生成甲基環戊烷完成整個反應。類似的,Petrocelli和Klein[36]使用硫化態Co-Mo/Al2O3催化劑,在壓力為7.0 MPa下發現酚和苯是二苯醚HDO反應的主要產物。在高的轉化率下(溫度高于300 ℃),酚被轉化為苯和環己烷(見圖9),這與Shabtai等人[37]的結果一致。

圖9 二苯醚的HDO反應機理Fig.9 HDO reaction mechanism of two phenyl ether
2.4酮類
酮類主要有兩種HDO反應途徑:(1)直接氫解生成烴類化合物;(2)先加氫生成醇,再氫解生成烴類化合物(見圖10)。

圖10 二苯甲酮的HDO反應途徑Fig.10 HDO reaction pathway of benzophenone
Durand等[38]在250 ℃下用Ni-Mo/γ-Al2O3催化劑催化環己酮加氫脫氧反應,環己烷為主要的反應產物,反應收率達95%。Oliva等[39]認為,在金屬中心催化環己酮C=O氫化生成環己醇,然后在酸中心脫水生成環己烯。不同載體對酮的加氫脫氧反應影響較大。Puente等[40]用硝酸對活性炭進行了改性處理,結果表明,在280 ℃,催化反應120 min,對甲基苯乙酮轉化率達100%,對甲基乙基苯為主要產物。
3 結語與展望
近年來,眾多研究者對模型含氧化合物加氫脫氧的機理等做了大量研究。但在含氧化合物的HDO反應過程中,由于采用的催化劑類型不同,反應途徑和產物的選擇性也有所不同。但是到目前為止,并未指出采用不同類型的催化劑所得產物選擇性有較大差異的本質原因。以后可從以下三個方面進行研究和改進:(1)進一步詳細研究含氧化合物在不同類型催化劑上的HDO反應機理,找到導致反應產物選擇性不同的本質原因;(2)開發新型的選擇性更好的適合煤焦油及模型化合物加氫脫氧催化劑;(3)通過大量含氧模型化合物HDO反應的研究,探究清楚實際油品中不同類型含氧化合物在HDO過程中的相互作用,為實際油品的HDO反應和工業化生產提供依據。
[1]Furimsky E. Catalytic hydrodeoxygenation[J]. Applied Catalysis A: General, 2000, 199(2): 147-190.
[2]Robert J A. An overview of modeling studies in HDS,HDN and HDO catalysis[J]. Polyhedron,1997, 16(18): 3073-3088.
[3]Ellott D C, Neuenschwander G G. Liquid fuels by low severity hydrotreating of biocrude[J]. Developments in Thermochemical Biomass Conversion, 1996, 1(2): 611-621.
[4]Grange P, Laurent E, Maggi R, et al. Hydrotreatment of pyrolysis oils from biomass: reactivity of the various categories of oxygenated compounds and preliminary techno-economical study [J]. Catal Today, 1996, 29(4): 297-301.
[5]Furimsky E. Chemistry of catalytic hydrodeoxygenation [J]. Catalysis Reviews: Science and Engineering, 1983, 25(3): 421-458.
[6]Grandy D W, Petrakis L, Young D C. Determination of oxygen functionalities in synthetic fuels by NMR of naturally abundant 17O[J]. Nature Y, 1984, 308(5955): 175-177.
[7]Petrakis L, Young D C, Ruberto R G. Catalytic hydroprocessing of SRC-Ⅱ heavy distillate fractions. 2.Detailed structural characterizations of the fractions[J]. Industrial and Engineering Chemistry Process Design and Development Y, 1983, 22(2): 298-305.
[8]Petrakis L, Ruberto R G, Young D C, et al. Catalytic hydroprocessing of SRC-Ⅱ heavy distillate fractions. 1.Preparation of the fractions by liquid chromatography[J]. Industrial and Engineering Chemistry Process Design and Development Y, 1983, 22(2): 292-298.
[9]耿層層,李術元,岳長濤,等.神木低溫煤焦油中含氧化合物的分析與鑒定[J].石油學報,2013,29(1):130-136.
[10]吳婷,凌鳳香,馬波,等.GC-MS分析低溫煤焦油酸性組分及堿性組分[J].石油化工離等學校學報,2013,26(3):44-52.
[11]Furimsky E. Mechanism of catalytic hydrodeoxygenation of tetrahydrofuran[J]. Industrial and Engineering Chemistry Product Research and Development A, 1983, 22(1): 31-34.
[12]Furimsky E. Deactivation of molybdate catalyst during hydrodeoxygenation of tetrahydrofuran[J]. Industrial and Engineering Chemistry Product Research and Development A, 1983, 22(1): 34-38.
[13]Furimsky E. The mechanism of catalytic hydrodeoxygenation of furan[J]. Applied Catalysis A, 1983, 6(2): 159-164.
[14]Bunch A Y, Ozkan U S. Investigation of the reaction network of benzofuran hydrodeoxygenation over sulfided and reduced Ni-Mo/Al2O3catalysts[J]. Journal of Ctalysis(Print) A, 2002, 206(2): 177-187.
[15]Bunch A Y, Wang X Q, Ozkan U S. Hydrodeoxygenation of benzofuran over sulfided and reduced Ni-Mo/γ-Al2O3catalysts: effect of H2S[J]. Journal of Molecular Catalysis A, Chemical A, 2007,270 (1-2): 264-272.
[16]Krishnamurthy S, Panvelker S, Shah Y T. Hydrodeoxygenation of dibenzofuran and related compounds[J]. AIChE Journal A, 1981, 27(6): 994-1001.
[17]Badilla O R, Pratt K C, Trimm D L. A study of nickel-molybdate coal-hydrogenation catalysts using model feedstocks[J]. Fuel A, 1979, 58(4): 309-314.
[18]Satterfield C N, Yang S H. Simultaneous hydrodenitrogenation and hydrodeoxygenation of model compounds in a trickle bed reactor[J]. Journal of Catalysis A, 1983, 81(2): 335-346.
[19]Lavopa V, Satterfield C N. Catalytic hydrodeoxygenation of dibenzofuran[J]. Energy and fuels A, 1987, 1(4): 323-331.
[20]Odebunmi E O, Ollis D F. Catalytic hydrodeoxygenation.1.Conversions of o-,p-,and m-cresols[J]. Journal of Catalysis, 1983, 80(1): 56-64.
[21]Echeandia S, Arias P L, Barrio V L, et al. Synergy effect in the HDO of phenol over Ni-W catalysts supported on active carbon:effect of tungsten precursors[J]. Applied Catalysis B, Environmental A, 2010, 101(1-2): 1-12.
[22]Gevert S B, Eriksson M, Eriksson P. Direct hydrodeoxygenation and hydrogenation of 2,6-and 3,5-dimethylphenol over sulphided CoMo catalyst[J]. Applied Catalysis A: General, 1994, 117(2): 151-162.
[23]Samchenko N P, Pavlenko N V. Reactivity of alkylphenols in liquid phase catalytic hydrogenation[J]. Reaction Kinetics and Catalysis Letters A, 1982, 18(1-2): 155-158.
[24]Shin E J, Keane M A, Catalytic hydrogen treatment of aromatic alcohols[J]. Journal of Catalysis (Print) A, 1998, 173(2): 450-459.
[25]Weigold H. Behaviour of Co-Mo-Al2O3catalysts in the hydrodeoxygenation of phenols[J].Fuel A,1982,61(10):1021-1026.
[26]Wandas R, Surygala J, Sliwka E. Conversion of cresols and naphthalene in the hydroprocessing of three-component model mixtures simulating fast pyrolysis tars[J]. Fuel (Guildford) A, 1996, 75(6): 687-694.
[27]Furimsky E. Catalytic hydrodeoxygenation[J]. Applied Catalysis A: General A, 2000, 199(2): 147-190.
[28]Vogelzang M W, Li C L, Schuit G C A, et al. Hydrodeoxygenation of 1-naphthol: activities and stabilities of molybdena and related catalysts[J]. Journal of catalysis (Print) A, 1983, 84(1): 170-177.
[29]徐春華.加氫脫氧反應對硫化態催化劑結構的影響[D].北京:石油化工科學研究院,2011.
[30]Senol O I, Ryymin E M, Viljava T R. Effect of hydrogen sulphide on the hydrodeoxygenation of aromatic and aliphatic oxygenates on sulphided catalysts[J]. Journal of Molecular Catalysis. A, Chemical A, 2007, 277(1-2): 107-112.
[31]Lin Y C, Li J L, Wan H P. Catalytic Hydrodeoxygenation of Guaiacol on Rh-Based and Sulfided CoMo and NiMo Catalysts[J]. Energy Fuels, 2011, 25(3): 890-896.
[32]王威燕,楊運泉,羅和安,等.La-Ni-Mo-B非晶態催化劑的制備加氫脫氧性能及失活研究[J].燃燒化學學報,2011,39(5):367-372.
[33]Bredenberg J B S, Huuska M, Toropainen P. Hydrogenolysis of differently substituted methoxyphenols[J]. Journal of Catalysis (Print) A. 1989, 120(2): 401-408.
[34]Kallury R K M R, Wanda M. Hydrodeoxygenation of hydroxy, methoxy, and methyl phenols with molybdenum oxide/nickel oxide/alumina catalyst[J]. Journal of Catalysis (Print) A, 1985, 96(2): 535-543.
[35]Artok L, Erbatur O, Schobert H H. Reaction of dinaphthyl and diphenyl ethers at liquefaction conditions[J]. Fuel processing technology A, 1996, 47(2): 153-176.
[36]Petrocelli F P, Klein M T. Modeling lignin liquefaction. I: Catalytic hydroprocessing of lignin-related methoxyphenols and interaromatic unit linkages[J]. Fuel Science and Technology International A, 1987, 5(1): 25-62.
[37]Shabtai J, Nag N K, Massoth F E. Catalytic functionalities of supported sulfides. IV: C-O hydrogenolysis selectivity as a function of promoter type[J]. Journal of Catalysis (Print) A, 1987, 104(2): 413-423.
[38]Durand R, Geneste P, Moreau C. Hetergeneous hydrodeoxygenation of ketones and alcohols on sulfided NiO-MoO3/γ-Al2O3catalyst[J]. Journal of Catalysis, 1984, 90(1): 147-149.
[39]Olivas A, Samano E C, Fuentes S. Hydrogenation of cyclohexanone on nickel-tungsten sulfide catalysts[J]. Applied Catalysis A:General, 2001, 220(1-2): 279-285.
[40]De L P, Gil G, Pis A J. Effects of support surface chemistry in hydrodeoxyg-enation reactions over CoMo/activated carbon sulfided catalysts[J]. Langmuir, 1999, 15(18): 5800-5806.
Research Progress on Carl Tar Hydrodeoxygenation
XURen-jun1,2,HUWei-yue3,CUIWen-gang4,LIDong4,LIWen-hong4
(1 Xi’an Institute of Light Industry,Shaanxi Xi’an 710001;2 Shaanxi Extend Oil Oilfield Chemical Technology Co., Ltd., Shaanxi Xi’an 716000; 3 Shaanxi Province Supervision and Inspection Institute of Product Quality,Shaanxi Xi’an 710048;4 School of Chemical Engineering, Northwest University,Shaanxi Xi’an 710069,China)
The types of the main oxygen-containing compounds in coal tar were briefly described, including phenols, naphthalene, esters, acids, aldehydes, furans and other oxygen-containing compounds, and the reaction mechanisms about different types of oxygen-containing compounds in hydrodeoxygenation(HDO) process was simply introduced. Meanwhile, reaction mechanism and pathway of different types of oxygen-containing compounds were also reviewed. The directly and indirectly in the process of HDO and path selection were summarized. For a complexity and diversity of oxygenated compounds contained in the coal tar, a number of improvements and new research directions were made. In addition, the prospect of oxygen-containing compounds HDO was discussed.
hydrodeoxygenation(HDO);oxygenated compound;reaction mechanism;kinetics
李穩宏(1955-),男,博導。
TQ517.4
A
1001-9677(2016)015-0039-05
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
汽車工程學報(2017年2期)2017-07-05 08:13:02
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
