1,1,2,3,4,5-六苯基硅雜環戊二烯X射線譜的理論研究
2016-09-05 13:04:08宋秀能王廣偉王傳奎山東師范大學物理與電子科學學院濟南250014
物理化學學報 2016年4期
宋秀能 王廣偉 常 燕 馬 勇 王傳奎(山東師范大學物理與電子科學學院,濟南250014)
1,1,2,3,4,5-六苯基硅雜環戊二烯X射線譜的理論研究
宋秀能王廣偉常燕馬勇*王傳奎
(山東師范大學物理與電子科學學院,濟南250014)
近年來苯基硅雜環戊二烯作為一類高效的有機發光二極管材料被廣泛研究。本工作利用密度泛函理論結合芯態空穴近似研究了1,1,2,3,4,5-六苯基硅雜環戊二烯分子中碳原子K殼層和硅原子L殼層的X射線光電子能譜和近邊X射線吸收精細結構譜,與實驗譜線符合較好。通過理論結果對實驗測量的1,1,2,3,4,5-六苯基硅雜環戊二烯分子的X射線譜進行了分析和標定。我們發現碳原子K殼層X射線光電子能譜在低能區283.8 eV處的譜峰是由于與硅原子成鍵的兩個電負性較強的碳原子導致的。碳原子K殼層近邊X射線吸收精細結構譜中最強的吸收峰與苯分子的吸收峰類似。硅原子L殼層近邊X射線吸收精細結構譜兩個主要吸收峰分別來自于σ*Si-C和π*Si-Ph躍遷。
苯基硅雜環戊二烯;X射線光電子能譜;近邊X射線吸收精細結構譜;密度泛函理論;完全芯態空穴
[Article]
www.whxb.pku.edu.cn
X射線光電子能譜(XPS)和近邊X射線吸收精細結構譜(NEXAFS)是研究分子化學結構和電子結構的有效工具,在表面科學領域具有廣泛應用8-10。X射線光譜技術可以通過探測材料中元素內層核軌道電子到連續態或未占據軌道的躍遷來研究原子的化學組成和化學環境,在有機材料的研究中具有重要作用。尤其是近邊X射線吸收精細結構譜由于其分辨率高同時具有極化相關特性,在探測有機太陽能電池材料11,12、有機發光二極管材料13以及多種多樣的自組裝單層膜等材料14的分子軌道信息和分子取向等信息的研究方面具有不可替代的作用。目前,X射線譜的研究主要集中在實驗方面,理論方面的研究較少。但是,理論研究工作在解釋實驗工作,尤其是解釋實驗測量中出現的無法標定的譜峰方面,具有特有的優勢。例如在石墨烯X射線吸收譜的實驗探測中,Pacilé15和Jeong16兩個實驗工作組對288 eV處譜峰的標定有著不同的解釋,Hua等17的理論工作對該爭議做了合理的解釋。對于另一種重要的功能分子材料石墨烯氧化物,Zhang等18對其X射線光電子能譜也進行了理論研究,其研究結果對于石墨烯氧化物X射線光電子能譜譜峰的標定及石墨烯的氧化過程均提供了準確的理論解釋。在氨表面的實驗測量中,氮原子1s軌道的X射線光電子能譜和近邊X射線吸收精細結構譜均出現未知譜峰9,本課題組對其進行了合理的理論解釋,得到了實驗工作者的認可14。總之,理論研究工作在對實驗結果的解釋和理解方面,具有極其重要的意義。
本工作中,我們研究了一種苯基硅雜環戊二烯分子:1,1,2,3,4,5-六苯基硅雜環戊二烯(HPS)的X射線光電子能譜和近邊X射線吸收精細結構譜。雖然一些實驗研究組已經利用X射線吸收光電子能譜和近邊X射線吸收精細結構譜對HPS分子做過一系列的研究13,19,但目前該方面的理論研究仍然較為少見。因此,從理論上對HPS分子的X射線譜進行研究和分析對于實驗光譜的理解是非常有意義的。我們在第一性原理的基礎上分別計算了碳原子1s軌道和硅原子2p軌道到未占據軌道和連續態的躍遷,對HPS分子的內層核軌道的電子結構以及X射線譜與電子結構、幾何結構之間的關系進行了深入的分析,對實驗譜線的解釋提供了理論依據。

圖1 HPS分子幾何結構Fig.1 Geometry structure of HPS molecule
2 理論模型和計算方法
1,1,2,3,4,5-六苯基硅雜環戊二烯的幾何結構如圖1所示。該分子的結構是利用包含長程修正項的M062X密度泛函20在Gaussian 09程序包21中完全放開所有原子進行優化的,計算中采用6-31G(d,p)基組22。同時在優化結構的基礎上進行了頻率分析,確保優化后得到的幾何結構為穩定的分子構型。在優化得到的穩定結構基礎上,利用密度泛函理論進行自然鍵軌道(NBO)電荷分析和X射線譜的計算。X射線譜是利用StoBe程序包23進行計算的,X射線譜計算方面采用的是Becke交換泛函24和Perdew關聯泛函25。在基函數的選擇上,我們選用IGLO-III基函數26來描述被激發的原子。在計算某個原子X射線譜的過程中,需要排除其他同類未激發原子的影響。因此對于未被激發的同類原子,采用有效核勢來模擬相應的核軌道。在該近似下,未被激發的碳原子僅考慮價軌道上的四個電子。對于其他所有原子則采用TZVP基函數27。
本文研究了兩種X射線譜:X射線光電子能譜和近邊X射線吸收精細結構譜。X射線光電子能譜對應電子從內層核軌道到電離態的激發,因此只需要計算內層核軌道的電離能(IP),將其能量和強度用高斯型函數展寬即可。內層核軌道電離能在數值上等于核電離態能量與基態(GS)能量之差。核電離態是一種能量非常高的激發態,該激發態的計算有很多方法。本文中利用了完全芯態空穴方法(FCH)28來計算該激發態的能量。與其他近似方法如等效芯態空穴(ECH)、激發芯態空穴(XCH)以及半芯態空穴(HCH)相比,該方法是目前在核激發態計算方面計算精度最高、與實驗測量結果符合最好的方法29。近邊X射線吸收精細結構譜則對應從內層核軌道到分子未占據軌道的躍遷。該譜線的計算是完全芯態空穴近似下采用ΔKohn-Sham (ΔKS)方法30,結合末態定則進行的31。近邊X射線吸收精細結構譜的吸收振子強度可以通過下式來計算:
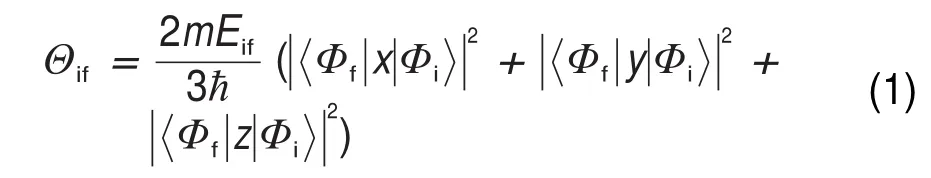
其中,Φi/f是X射線吸收過程中涉及到的兩個分子軌道(i表示初態,f表示末態),Θif是從Φi到Φf的吸收振子強度,m是電子質量,Eif是兩個軌道的能級間隔,?是約化普朗克常數。系數1/3表示在x、y、z三個方向取平均而得到統計結果,體現分子在聚集態中隨機排布的分子取向。得到躍遷能與振子強度之后我們將利用精確的第一激發能,也就是從1s(2p)軌道到LUMO的躍遷能來標定整個譜線。然后將標定好的譜線進行展寬即可得到最終譜線。對于譜線中低于電離能的范圍,利用高斯型函數進行展寬,而高于電離能的范圍,也就是連續態范圍譜線的展寬采用的是Stieltjes方法32。本工作中X射線光電子能譜的半高全寬采用的是0.3 eV,近邊X射線吸收精細譜低能區半高全寬為0.6 eV。由于自旋與軌道相互作用,硅原子的2p軌道會劈裂為2p1/2和2p3/2兩個軌道。我們計算X射線譜利用的StoBe程序無法描述此相互作用,因此本文中利用近似方法處理。即利用計算的2px軌道能量(與2py,2pz能量相同)作為2p1/2軌道的能量,而2p3/2軌道則在2p1/2軌道能量基礎上紅移0.6 eV,同時其強度比為1:2,該能量平移值與強度比取自SiO2譜線的實驗值33。
3 結果與討論
3.1X射線光電子能譜
我們利用M062X泛函20計算了HPS分子的幾何結構,該泛函考慮了苯環之間的弱相互作用使得到的幾何結構更為合理。HPS分子中的某些苯環可以自由轉動,因此存在較多可能的分子構型。但由于X射線譜主要與原子的局域化學環境有關,因此苯環扭轉角的不同對X射線譜的影響不大。本工作中,我們選取的分子構型是在相同計算層次下相對能量最低的構型。在這個幾何構型的基礎上,計算了碳原子和硅原子的X射線譜。我們首先計算了HPS分子中碳原子1s軌道和硅原子2p軌道的X射線光電子能譜。根據對稱性分析可知,HPS分子中有20個不等價碳原子,其中有兩個不等價碳原子位于戊二烯雜環中,其余不等價碳原子全部位于苯環上。表1中給出了理論計算的所有不等價碳原子1s軌道和硅原子2p軌道的電離能。圖2分別給出了HPS分子中碳原子1s軌道和硅原子2p軌道實驗測量以及理論計算得到的X射線光電子能譜。圖中的實驗數據來源于Diller等13的工作。實驗測量的HPS分子碳原子1s軌道的X射線光電子能譜的主峰位置在大約284.8 eV附近,同時實驗測量得到的HPS分子碳原子1s軌道的X射線光電子能譜在低能區有一個不明顯的肩峰存在。通過對兩種譜線的對比,我們發現理論結果與實驗結果符合較好。理論計算的HPS分子中碳原子1s軌道的X射線光電子能譜的主峰位置在284.8 eV。通過分析所有不等價碳原子1s軌道的電離能可以看出,碳原子1s軌道的X射線光電子能譜的主峰是由苯環上的碳原子以及硅雜環中不與硅原子成鍵的兩個碳原子(C3/C4)貢獻的,而與硅原子成鍵的兩個碳原子(C1/C2)是HPS分子X射線光電子能譜譜峰中出現在283.8 eV處肩峰的原因。我們對HPS分子進行了NBO電荷分析,HPS分子中的硅原子帶有+1.68e的正電荷,與硅原子相連的兩個碳原子各帶有-0.34e的負電荷,該結果與實驗預測的結果非常符合。與硅原子相連的兩個碳原子與其他碳原子相比,具有較大的電負性,原子電負性的增強會降低內殼層軌道的電離能。HPS分子中大部分位于苯環中的碳原子化學環境類似,均與碳原子或氫原子成鍵,因此其1s軌道的電離能較為接近,該類碳原子1s軌道電離能集中在284.4 eV到285.0 eV范圍。HPS分子中有兩個苯環與硅原子相連,苯環中與硅原子相連的兩個碳原子1s軌道的電離能約在284.4 eV。

表1 理論計算的所有不等價C原子1s軌道和Si原子2p軌道電離能Table 1 Calculated ionization potentials of all nonequivalent carbon 1s and silicon 2p orbitals

圖2 理論計算與實驗測量的HPS分子中C原子1s軌道及Si原子2p軌道的X射線光電子能譜Fig.2 Theoretical and experimental C 1s and Si 2p X-ray photoelectron spectroscopy of HPS molecule
圖2(b)所示的是HPS分子中硅原子2p軌道實驗測量以及理論計算得到的X射線光電子能譜。實驗測量的HPS分子中硅原子2p軌道的X射線光電子能譜顯示,HPS中硅原子的2p1/2和2p3/2的電離能大約在100.6和101.3 eV。本工作中理論計算的HPS分子中硅原子2p軌道的電離能與實驗符合較好。理論計算的HPS中硅原子2p1/2和2p3/2軌道的電離能分別在100.6和101.2 eV。通過與實驗譜線的對比,可以發現理論計算的單分子的X射線光電子能譜與實驗測量的以聚集態存在的分子薄膜的譜線非常吻合。首先,這與X射線譜本身的特點有關,X射線譜是與內殼層軌道相關的譜線,主要表現原子自身的性質以及原子所處的局域化學環境。對于聚集態存在的分子薄膜,雖然分子間存在相互作用,但由于該相互作用為分子間的弱相互作用,因此對X射線譜的影響不大;其次,理論譜線與實驗譜線的吻合再次證明本工作中理論方法的合理性,可以利用理論計算結果對實驗譜線進行對比和解釋。
3.2近邊X射線吸收精細結構譜
與X射線光電子能譜相比,近邊X射線吸收精細結構譜的分辨率更高,而且能夠給出分子未占據軌道的信息,因此該譜線在實驗研究中應用更為廣泛。圖3中給出的是理論計算的HPS分子中碳原子1s軌道和硅原子2p軌道的近邊X射線吸收精細結構譜與實驗譜線的對比。近邊X射線吸收精細結構譜是一種極化相關的譜線,X射線的極化方向和分子軌道方向的夾角不同會導致譜峰強度的變化,因此通過改變入射X射線與樣品之間的角度可以測量樣品表面分子的分子取向34。Diller等13在實驗中分別測量了7°和90°兩種入射角度的近邊X射線吸收精細結構譜用以分析HPS分子的分子取向。然而,HPS分子中的苯環為放射狀排布,而放射狀分子結構會導致分子軌道的無序性,即使該分子在薄膜中有序排列,也無法通過極化的近邊X射線吸收精細結構譜來判斷其分子取向。因此,實驗測量的當X射線以7°與90°入射時得到的近邊X射線吸收精細結構譜差別較小。本工作中,我們采用的是公式(1)對近邊X射線吸收精細結構譜進行計算,該公式對應于入射角為55°時測得的譜線,等效于分子隨機排布時測量的譜線34。
從圖3(a)可以看出,HPS分子碳原子1s軌道的實驗譜線中有四個較為明顯的譜峰,依次為283.7 eV(I)、285.1 eV(II)、287.6 eV(III)和289.0 eV (IV)。通過與實驗譜線的對比,我們發現理論計算的HPS分子碳原子1s軌道近邊X射線吸收精細結構譜與實驗測量的譜線整體符合較好,理論計算得到的譜峰位置及其相對強度都較為符合,可以通過理論結果對實驗譜峰進行詳細的分析和準確的標定。表2給出了理論計算的碳原子1s軌道和硅原子2p軌道近邊X射線吸收精細結構譜中主要躍遷的躍遷能及躍遷強度。理論計算結果較準確地在284.9 eV處擬合了非對稱吸收峰II,該吸收峰的主峰伴隨著一個低能區以及一個高能區的肩峰。根據理論分析可知,II峰是由于苯環中的碳原子1s軌道到LUMO軌道的躍遷,由于HPS分子中有六個苯環,因此該吸收峰的強度遠大于其他三個共振吸收峰。HPS分子碳原子1s軌道近邊X射線吸收精細結構譜在低能區的283.7 eV處有一個強度較弱的吸收峰,該吸收峰是由于戊二烯環中碳原子1s軌道到LUMO軌道的躍遷,由于該類碳原子數目較少,因此該躍遷的強度較小。實驗譜線中高能區的譜峰由于分辨率限制無法分辨,而理論計算的譜峰則可以較為明顯地看到。根據譜線分析可以知道,該高能區強度較弱的譜峰是由于與C1/ C2兩個碳原子相連的苯環上碳原子1s軌道到LUMO+1軌道躍遷的貢獻。對于實驗譜中的287.6和289.0 eV兩個位置的共振吸收峰,我們計算模擬的共振吸收的位置為287.8和289.1 eV,與實驗符合較好。在287.8 eV處的吸收峰主要是由于HPS分子六個苯環上的碳原子1s軌道到LUMO+ 3、LUMO+4軌道的共振躍遷。而289.1 eV處的吸收峰主要是由于與硅原子相連的兩個苯環中碳原子1s軌道到LUMO+4軌道的躍遷。

圖3 理論計算與實驗測量的HPS分子中C原子1s軌道和Si原子2p軌道近邊X射線吸收精細結構譜Fig.3 Theoretical and experimental C 1s and Si 2p near-edge X-ray absorption fine structure spectroscopy of HPS molecule

表2 理論計算的C原子1s軌道和Si原子2p軌道近邊X射線吸收精細結構譜主要吸收峰的躍遷能及躍遷強度Table 2 Calculated energies and intensities of major transitions in C 1s and Si 2p near-edge X ray absorption fine structure spectroscopy
圖3(b)給出了理論計算和實驗測量的硅原子2p軌道的近邊X射線吸收精細結構譜。與碳原子1s軌道近邊X射線吸收精細結構譜類似,當X射線以7°和90°入射時實驗測量的硅原子2p軌道的近邊X射線吸收譜差別不大。實驗譜線中主要有兩個吸收峰,分別位于102.6 eV(V)和106.2 eV(VI),同時,這兩個主峰的左側各有一個肩峰,分別位于100.7和104.9 eV附近。由于自旋與軌道相互作用的影響,硅原子2p軌道的近邊X射線吸收譜的計算與碳原子1s軌道的譜線計算相比更加困難。一般來講,電子和電子之間的相互作用是非常難預測的,目前處理該問題的一個方法是利用多重散射理論35,36。由于本工作重點在于標定不同碳原子1s軌道的近邊X射線吸收精細結構譜,同時本工作使用的StoBe程序包未包含較精確的處理自旋與軌道相互作用的成分,因此對于硅原子2p軌道的近邊X射線吸收精細結構譜,我們利用的是與計算X射線光電子能譜同樣的近似方法。理論計算的硅原子2p軌道近邊X射線吸收精細結構譜在100.8、101.4、102.7和105.0 eV處各有一個較強的吸收峰。理論計算的譜線與實驗測量的譜線相比,譜峰位置和譜峰相對強度基本符合。理論計算的100.8、101.4和102.7 eV處的共振吸收是由于2p軌道到LUMO、LUMO+2、LUMO+3軌道的躍遷導致,實驗譜線中102.6 eV處的V峰應該包含以上三種躍遷成分。理論計算的105.0 eV處較強的共振吸收是由于2p軌道到LUMO+13軌道的躍遷導致,該譜峰位置與實驗測量的VI峰的肩峰位置接近,然而我們更相信該譜峰對應于實驗譜線中的VI主峰。圖4給出了上述幾個共振吸收對應的末態分子軌道,從圖中可以明顯地看出LUMO軌道屬于典型的π*軌道,LUMO+2和LUMO+3軌道則是σ*軌道,而LUMO+13又是π*軌道。Urquhart等37在其他硅化合物的近邊X射線譜測量時將V峰和VI峰標定為σ*Si-C和σ*Si-O,我們根據理論軌道成分分析后認為實驗測量的HPS分子中硅2p軌道近邊X射線吸收精細結構譜中的VI峰為π*Si-Ph躍遷;而實驗測量的譜線中的V峰,雖然包含2p軌道到LUMO的共振吸收,但考慮其貢獻不大,因此我們認為該吸收峰應該為σ*Si-C躍遷。從圖3可以看出理論計算的譜線中各個譜峰的相對強度與實驗譜線相比并不完全吻合,這與理論工作中利用到的一個近似方法有關。理論計算的譜峰強度與共振吸收對應的未占據軌道波函數有關。而在實際的實驗測量過程中,譜峰強度是與內層電子被激發后的電子態有關。利用一個分子軌道來近似地模擬一個電子態,會導致計算的振子強度出現差異。因此,我們計算的主峰和肩峰的相對強度無法與實驗完全匹配。這是目前使用該近似時不可避免的。另外,由于共振吸收峰較多,理論模擬時采用不同的半高全寬也會導致譜峰相對強度的不同。

圖4 理論計算的HPS分子未占據軌道Fig.4 Theoretical unoccupied orbitals of HPS molecule
4 結論
我們在完全芯態空穴近似下,利用密度泛函理論研究了1,1,2,3,4,5-六苯基硅雜環戊二烯分子中碳原子1s軌道和硅原子2p軌道的X射線光電子能譜和近邊X射線吸收精細結構譜。理論計算得到的譜線與已有實驗譜線符合較好。通過對碳原子1s軌道和硅原子2p軌道的X射線譜的理論分析,我們對實驗譜線中出現的譜峰分別進行了標定。碳原子1s軌道X射線光電子能譜中的低能區的肩峰是由于與硅原子相連的兩個碳原子貢獻的,該類碳原子由于與硅原子成鍵而具有較大的電負性,從而導致較低的電離能。硅原子2p軌道X射線光電子能譜與實驗符合較好,同時NBO分析顯示硅原子處在典型的陽離子狀態,帶有+1.68e的電荷,而與其相毗鄰的碳原子則具有較強的電負性。理論計算的近邊X射線吸收精細結構譜與實驗測量的譜線在譜峰位置和譜峰強度方面都比較符合。我們對碳原子1s軌道和硅原子2p軌道的近邊X射線吸收精細結構譜進行了標定。碳原子1s軌道的吸收譜在285.1 eV處的吸收峰是由于HPS分子中苯環上的碳原子貢獻的。硅原子2p軌道的吸收譜存在兩個主要的吸收峰,分別被標定為σ*Si-C和π*Si-Ph躍遷。
References
(1)Ma,Q.Y.;Guan,R.F.;Li,G.Z.;Feng,S.Y.Chin.J.Org. Chem.2011,31(9),1395.[馬慶宇,關瑞芳,李國忠,馮圣玉.有機化學,2011,31(9),1395.]
(2)Mao,L.Y.;Wan,J.H.;Li,Z.F.;Tao,L.;Qiu,H.Y.Prog. Chem.2009,21(10),2153.
[毛林燕,萬俊華,李志芳,陶蘭,邱化玉,化學進展,2009,21(10),2153.]
(3)Deng,C.M.;Niu,Y.L.;Peng,Q.;Shuai,Z.G.Acta Phys.-Chim.Sin.2010,26(4),1051.
[鄧春梅,牛英利,彭謙,帥志剛.物理化學學報,2010,26(4),1051.] doi:10.3866/PKU.WHXB20100420
(4)Luo,J.;Xie,Z.;Lam,J.W.Y.;Cheng,L.;Chen,H.;Qiu,C.; Kwok,H.S.;Zhan,X.;Liu,Y.;Zhu,D.;Tang,B.Z.Chem. Commun.2001,1740.doi:10.1039/B105159H
(5)Braye,E.H.;Hübel,W.Chem.Ind.(London)1959,1250.
(6)Braye,E.H.;Hübel,W.;Caplier,I.J.Am.Chem.Soc.1961, 83,4406.doi:10.1021/ja01482a026
(7)Zhang,T.;Jiang,Y.;Niu,Y.;Wang,D.;Peng,Q.;Shuai,Z.J. Phys.Chem.A 2014,118,9094.doi:10.1021/jp5021017
(8)St?hr,J.NEXAFS Spectroscopy;Springer Verlag:Berlin,1996; pp 1-3.
(9)Dietrich,P.M.;Graf,N.;Gross,T.;Lippitz,A.;Krakert,S.; Schupbach,B.;Terfort,A.;Unger,W.E.S.Surf.Interface Anal.2010,42,1184.doi:10.1002/sia.v42:6/7
(10)Baio,J.E.;Weidner,T.;Brison,J.;Graham,D.J.;Gamble,L. J.;Castner,D.G.J.Electron Spectrosc.Relat.Phenom.2009, 172,2.doi:10.1016/j.elspec.2009.02.008
(11)Song,X.;Hua,W.;Ma,Y.;Wang,C.;Luo,Y.J.Phys.Chem.C 2012,116,23938.doi:10.1021/jp307834x
(12)Ma,Y.;Wang,G.W.;Sun,S.T.;Song,X.N.Acta Phys.-Chim.Sin.2015,31(8),1483.
[馬勇,王廣偉,孫紹濤,宋秀能.物理化學學報,2015,31(8),1483.] doi:10.3866/PKU.WHXB201505251
(13)Diller,K.;Ma,Y.;Luo,Y;Allegretti,F.;Liu,J.;Tang,B.Z.; Lin,N.;Barth,J.V.;Klappenberger,F.Phys.Chem.Chem. Phys.2015,17,31117.doi:10.1039/C5CP02935J
(14)Song,X.;Ma,Y.;Wang,C.;Dietrich,P.D.;Unger,W.E.S.; Luo,Y.J.Phys.Chem.C 2012,116(23),12649. doi:10.1021/jp302716w
(15)Pacilé,D.;Papagno,M.;Fraile,R.A.;Grioni,M.;Papagno,L. Phys.Rev.Lett.2008,101,066806.doi:10.1103/ PhysRevLett.101.066806
(16)Jeong,H.K.;Noh,H.J.;Kim,J.Y.;Colakerol,L.;Glans,P. A.;Jin,M.H.;Smith,K.E.;Lee,Y.H.Phys.Rev.Lett.2009, 102,099701.doi:10.1103/PhysRevLett.102.099701
(17)Hua,W.;Gao,B.;Li,S.;?gren,H.;Luo,Y.Phys.Rev.B 2010, 82,155433.doi:10.1103/PhysRevB.82.155433
(18)Zhang,W.H.;Carravetta,V.;Li,Z.Y.;Luo,Y.;Yang,J.L. J.Chem.Phys.2009,131,244505.doi:10.1063/1.3276339
(19)Dong,L.;Wang,W.;Lin,T.;Diller,K.;Barth,J.V.;Liu,J.; Tang,B.Z.;Klappenberger,F.;Lin,N.J.Phys.Chem.C 2015, 119,3857.doi:10.1021/acs.jpcc.5b00116
(20)Zhao,Y.;Truhlar,D.G.Accounts Chem.Res.2008,41,157. doi:10.1021/ar700111a
(21)Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 09,Revision D.01;Gaussian Inc.:Wallingford,CT,2009.
(22)Rassolov,V.;Pople,J.A.;Ratner,M.;Windus,T.L.J.Chem. Phys.1998,109(4),1223.doi:10.1063/1.476673
(23)Hermann,K.;Pettersson,L.;Casida,M.;et al.StoBe,Version 3.0;StoBe Software:Stockholm,Sweden,2007.
(24)Becke,A.D.Phys.Rev.A 1988,38,3098.doi:10.1103/ PhysRevA.38.3098
(25)Perdew,J.P.Phys.Rev.B 1986,33,8822.doi:10.1103/ PhysRevB.33.8822
(26)Kutzelnigg,W.;Fleischer,U.;Schindler,M.NMR:Basic Principles and Progress;Springer Verlag:Berlin Heidelberg, 1990;Vol.213.
(27)Sch?fer,A.;Huber,C.;Ahlrichs,R.J.Chem.Phys.1994,100 (8),5829.doi:10.1063/1.467146
(28)Nyberg,M.;Luo,Y.;Triguero,L.;Pettersson,L.G.M.;?gren, H.Phys.Rev.B 1999,60,7956.doi:10.1103/ PhysRevB.60.7956
(29)Hua,W.J.;Gao,B.;Luo,Y.Prog.Chem.2012,24(6),964.
[花偉杰,高斌,羅毅,化學進展,2012,24(6),964.]
(30)Triguero,L.;Pettersson,L.G.M.;?gren,H.Phys.Rev.B 1998,58,8097.doi:10.1103/PhysRevB.58.8097
(31)von Barth,U.;Grossman,G.Phys.Rev.B 1982,25,5150.doi: 10.1103/PhysRevB.25.5150
(32)Triguero,L.;Plashkevych,O.;Pettersson,L.G.M.;?gren,H. J.Electron Spectrosc.Relat.Phenom.1999,104(1-3),195. doi:10.1016/S0368-2048(99)00008-0
(33)Li,D.;Bancroft,G.M.;Kasrai,M.;Fleet,M.E.;Secco,R.A.; Feng,X.H.;Tan,K.H.;Yang,B.X.Am.Min.1994,79,622.
(34)Song,X.;Wang,G.;Ma,Y.;Jiang,S.;Yue,W.;Xu,S.;Wang, C.Chem.Phys.Lett.2016,645,164.doi:10.1016/j. cplett.2015.12.005
(35)Chaboy,J.;Benfatto,M.;Davoli,I.Phys.Rev.B 1995,52, 10014.doi:10.1103/PhysRevB.52.10014
(36)Xiong,J.Z.;Jiang,D.T.;Liu,Z.F.;Baines,K.M.;Sham,T. K.;Urquhart,S.G.;Wen,A.T.;Tyliszczak,T.;Hitchcock,A. P.Chem.Phys.1996,203,81.doi:10.1016/0301-0104(95) 00353-3
(37)Urquhart,S.G.;Turci,C.C.;Tyliszczak,T.;Brook,M.A.; Hitchcock,A.P.Organometallics 1997,16,2080.doi:10.1021/ om961028f
Theoretical Study on X-Ray Spectroscopy of 1,1,2,3,4,5-Hexaphenylsilole
SONG Xiu-NengWANG Guang-WeiCHANG YanMAYong*WANG Chuan-Kui
(School of Physics and Electronics,Shandong Normal University,Jinan 250014,P.R.China)
As an effective organic light-emitting diode,the benzene-based silole has recently been widely researched.We calculate the carbon K edge and silicon L edge X-ray photoelectron spectroscopy and nearedge X-ray absorption fine structure spectroscopy of the 1,1,2,3,4,5-hexaphenylsilole(HPS)molecule with density functional theory.The theoretical results match the available experimental spectra very well.The experimental X-ray spectra were analyzed and assigned by our theoretical results.It is found that the peak at 283.8 eV in the carbon K edge X-ray photoelectron spectroscopy is caused by the two carbon atoms bonding with the silicon atom.The carbon K edge near-edge X-ray absorption fine structure spectroscopy possesses a strong resonance absorption similar with that observed for a benzene molecule.The two main resonances in silicon L edge near-edge X-ray absorption fine structure spectroscopy were assigned to σ*Si-Cand π*Si-Phtransitions.
Benzene-based silole;X-ray photoelectron spectroscopy;Near-edge X-ray absorption fine structure spectroscopy;Density functional theory;Full core hole
1 引言
硅雜環戊二烯(又名硅咯、噻咯)是環戊二烯中橋頭碳原子被硅原子取代后形成的五元雜環。由于硅原子的σ*軌道與丁二烯中π*軌道存在特殊的σ*-π*共軛作用,使得該五元雜環的最低未占據分子軌道(LUMO)遠低于其他五元雜環,意味著該材料具有更加優秀的電子親和性質1-3。該性質使其成為光電領域尤其是有機發光二極管方面的一種理想材料。硅雜環戊二烯材料的另一個重要特性是其具有與其他大部分發光材料聚集猝滅性質(ACQ)相反的聚集誘導(AIE)性質4,在聚集狀態下有熒光增強現象。近幾十年來,人們一直利用不同的官能團來調節硅雜環戊二烯的電子結構,改善其光學性能。其中,利用一定數量的苯環對該五元雜環修飾而形成的苯基硅雜環戊二烯是第一類被合成的硅雜環戊二烯衍生物5,6。基于苯基硅雜環戊二烯的分子器件作為一類高效的有機發光二極管材料被廣泛關注,其熒光量子產率、電荷載流子傳輸性質以及最高占據分子軌道(HOMO)和LUMO方面的性質是現階段研究的重點內容7。然而到目前為止,對苯基硅雜環戊二烯分子內層核軌道層次光譜的理論研究較為少見。
November 25,2015;Revised:January 28,2016;Published on Web:January 29,2016.*Corresponding author.Email:mayong@sdnu.edu.cn;Tel:+86-13793154301. The project was supported by the National Natural Science Foundation of China(21303096,11374195),Promotive Research Fund for Young and Middle-Aged Scientists of Shandong Province,China(BS2013CL016,BS2014CL039),China Postdoctoral Science Foundation(2013M541951, 2014T70663),Scientific Research Foundation for the Returned Overseas Chinese Scholars,State Education Ministry,and Program of Domestic Study for Young Scholar of University in Shandong Province Sponsored by Shandong Provincial Educational Department,China.
O641
10.3866/PKU.WHXB201601291
國家自然科學基金(21303096,11374195),山東省優秀中青年科學家科研獎勵基金(BS2013CL016,BS2014CL039),中國博士后科學基金(2013M541951,2014T70663),教育部留學回國人員科研啟動基金和山東省高等學校青年骨干教師國內訪問學者項目資助
猜你喜歡
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
當代陜西(2022年5期)2022-04-19 12:10:18
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
新世紀智能(數學備考)(2021年9期)2021-11-24 01:14:28
湘潮(上半月)(2021年4期)2021-07-20 08:05:28
汕頭大學學報(自然科學版)(2020年4期)2020-12-14 07:05:00
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
學習月刊(2015年21期)2015-07-11 01:51:44
