茶樹咖啡堿合成酶CRISPR/Cas9基因組編輯載體的構建
2016-09-21 09:51:57唐雨薇劉麗萍王若嫻陳宇宏劉仲華劉碩謙
茶葉科學 2016年4期
唐雨薇,劉麗萍,王若嫻,陳宇宏,劉仲華,2,3,劉碩謙,2,3*
1.湖南農業大學園藝園林學院,湖南 長沙 410128;2.國家植物功能成分利用工程技術研究中心,湖南 長沙 410128;3.教育部茶學重點實驗室,湖南 長沙 410128
茶樹咖啡堿合成酶CRISPR/Cas9基因組編輯載體的構建
唐雨薇1,3,劉麗萍1,3,王若嫻1,陳宇宏1,劉仲華1,2,3,劉碩謙1,2,3*
1.湖南農業大學園藝園林學院,湖南 長沙 410128;2.國家植物功能成分利用工程技術研究中心,湖南 長沙 410128;3.教育部茶學重點實驗室,湖南 長沙 410128
CRISPR/Cas9技術是一門新興的基因組定點編輯技術,具有操作簡單、高效的優點,可輕松實現對目標基因的敲除、替換和定點突變等操作。該技術剛誕生,就受到了全球生命科學領域研究者的關注,不到 3年的時間就已經成功應用于多種動、植物當中。然而 CRISPR/Cas9技術在茶樹中的應用面臨載體構建問題,本文以茶樹咖啡堿合成酶為例,聯合采用常規PCR、Overlapping PCR和Golden Gate Cloning技術,構建了包含茶樹咖啡堿合成酶雙靶點的CRISPR/Cas9基因編輯載體,為CRISPR/Cas9介導的基因組編輯技術在茶樹中的應用奠定了堅實基礎。
茶樹;基因組編輯技術;咖啡堿合成酶;CRISPR/Cas9技術
雖然我國一直非常重視茶樹的品種改良工作,但由于茶樹具有生長周期長、自交不親和的特性,使得采用常規育種技術難以實現茶樹育種工作取得突破性進展。近年來分子育種及基因工程等現代分子生物學技術在水稻、西紅柿、馬鈴薯等農作物品種改良中發揮了重要作用,也為茶樹育種提供了新的途徑。因而,茶樹分子生物學研究成了茶葉科學中最活躍和進展最快的領域。最近,茶樹轉錄組測序工作取得了重要進展[1-4]。目前,GenBank已收錄了約35萬條茶樹mRNA序列(http://www.ncbi.nlm.nih.gov/nuccore/?term=Camellia+ sinensis),幾乎涵蓋了茶樹中所有功能基因編碼序列。此外,我國茶樹的全基因組測序工作也于幾年前在多家單位開始啟動,并已相繼完成,茶樹全基因組序列將會在短期內釋放[5]。然而,面對大量的基因序列,我們對其功能了解卻是非常缺少,制約了基因資源的利用。因此,確定茶樹新基因的功能并高效利用新基因將是未來茶樹分子生物學研究中一個非常重要的主題,也就是說茶樹后基因組時代即將來臨。隨著現代分子生物技術的突飛猛進,新型基因組編輯技術CRISPR/Cas9(Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR-associated protein 9)將會在茶樹功能基因研究中發揮重要作用,預計將成為茶樹后基因組時代不可或缺的研究工具。
基因組定點編輯技術是分子育種、基因的功能研究和遺傳改造等重要工具,是通過某種途徑對基因組DNA特定位點進行改造的一種手段。迄今為止,應用時間比較長的基因組編輯技術有鋅指核酸酶(Zinc-finger nucleases,ZFN)[6]和類轉錄激活因子效應物核酸(Transcription activator-likeeffector nucleases,TALEN)技術[7]。但由于這兩項技術DNA結合結構域的改造較為復雜,對每一個基因位點的編輯都需要重新設計、合成和組裝2個核酸酶,載體構建困難,成為限制其發展的瓶頸[8]。科學家一直在尋找精確而簡單的基因組編輯方法,直到 2013年發現了CRISPR/Cas9技術[9]。CRISPR/Cas9技術是通過一段RNA來識別靶位點,因而在設計和構建上更加簡單,只需合成一個sgRNA就能實現對基因的特異性編輯[10]。自 CRISPR/Cas9技術的誕生到現在,短短幾年內該技術迅速應用于世界各地的實驗室,并成為基因組定點編輯的首選方法[11]。
CRISPR/Cas廣泛存在于古生菌和細菌的基因組中,屬自身免疫系統,可降解入侵的病毒或質粒 DNA[12]。在該免疫系統中,Cas蛋白(CRISP-associated protein)含有兩個核酸酶結構域,可以分別切割兩條DNA鏈,切割后DNA雙鏈斷裂從而使入侵的外源DNA降解[13]。CRISPR/Cas系統的組成結構比較固定,由 CRISPR序列與 Cas基因家族組成,其中CRISPR由一系列高度保守的重復序列(Repeat)與間隔序列(Spacer)相間排列組成,間隔序列可特異識別外源 DNA[13]。在CRISPR序列附近存在高度保守的CRISPR相關基因,這些基因編碼的蛋白具有核酸酶功能,可以對DNA序列進行特異性切割[14]。
CRISPR系統分為3種類型,現在常用的CRISPR/Cas系統由類型Ⅱ系統改造而來。類型Ⅱ系統的特征性蛋白為Cas9蛋白,具有加工產生 CRISPR RNA(crRNA)和切割雙鏈DNA功能。crRNA(CRISPR-derived RNA)通過堿基配對與 tracrRNA(trans-activating RNA)結合形成tracrRNA/crRNA復合體,此復合體一旦形成就能引導核酸酶Cas9蛋白在與crRNA配對的序列靶位點剪切雙鏈DNA。而tracrRNA/crRNA復合體可通過人工設計,融合crRNA與tracrRNA形成sgRNA(single-guided RNA),sgRNA足以引導Cas9 對DNA的定點切割(圖1)[13,15]。sgRNA可以通過載體表達或者化學合成后與Cas9蛋白共同進入細胞,對特異DNA序列剪切,從而促使DNA 發生NHEJ(nonhomologousend-joining)導致的基因缺失或同源重組,實現基因敲除[16]。CRISPR-Cas9 體系的RNA-DNA識別機制為選擇性基因組編輯提供了一個簡便而強大的工具。來自Streptococcus pyogenes的Cas9由于識別序列僅為2個堿基(GG),幾乎可以在所有的基因中找到大量靶點,因此得到廣泛的應用。Cas9蛋白在目前測試過的幾乎所有生物和細胞中均有活性,包括細菌、酵母、植物、魚,以及哺乳動物細胞[17]。該體系其中一個最重要的優勢是Cas9蛋白可在多個不同的sgRNA的引導下同時修飾多個基因組靶點。
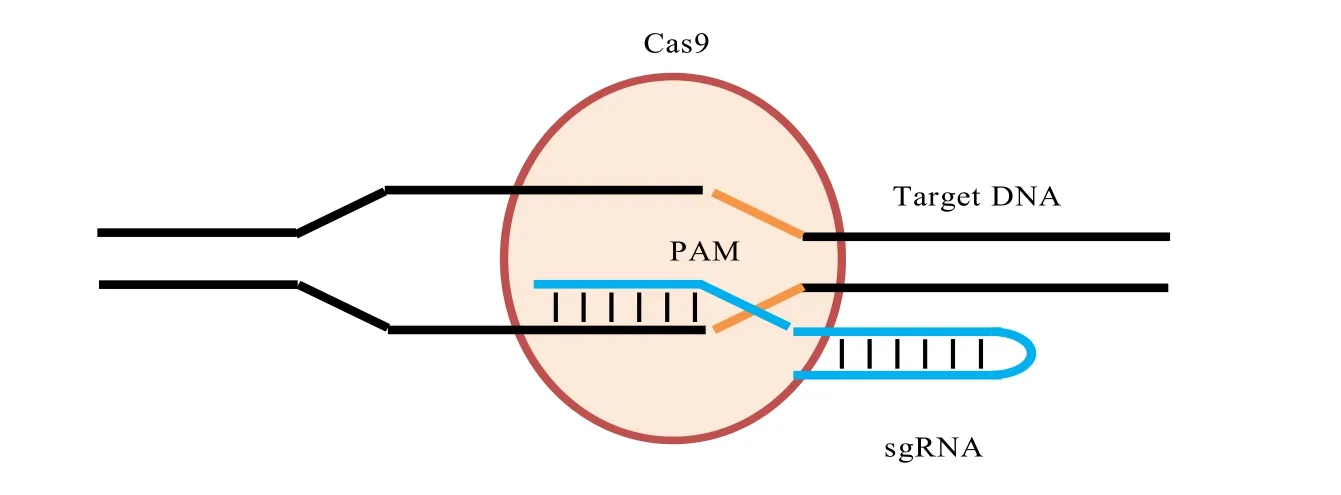
圖1 CRISPR/Cas9工作原理[15]Fig.1 The working model of the CRISPR/Cas9 system[15]
目前植物CRISPR/Cas9系統已日趨完善,已在十多種植物中成功實現了定點基因組編輯,除了在本氏煙草[18-19]、擬南芥[20]等模式植物上獲得了成功,很快在小麥[21]、玉米[22]、水稻[23]、高粱[19]、西紅柿[24]以及甜橙[25]等農作物上也成功實現了 CRISPR/Cas9的應用。最近,先后利用 CRISPR/Cas9技術實現了木本植物楊樹[26]和觀賞植物牽牛花[27]基因組的定點編輯,將 CRISPR/Cas9系統在植物中應用推上了新的領域。
然而,到目前為止尚未見有關CRISPR/Cas9技術在茶樹上的應用報道,其中一個重要的原因是針對茶樹的 CRISPR/Cas9基因編輯載體構建技術尚不完善。本文以茶樹中咖啡堿合成酶為例,建立茶樹CRISPR/Cas9基因編輯載體的構建方法,為 CRISPR/Cas9基因編輯技術在茶樹中的應用奠定理論基礎。
1 材料與方法
1.1質粒、引物、菌種與試劑
植物 CRISPR/Cas9基因編輯載體為pYLCRISPR/Cas9P35S-H,GenBank登錄號為AY310901,是在雙元載體質粒pCAMBIA1300(AF234296)的基礎上進行改造而來的,由花椰菜花葉病毒(CaMV)35 S啟動子驅動Cas9p序列的表達。該質粒在E.coli TOP10F′(LacIq)菌株繁殖,該菌株LacIq基因型產生的阻礙蛋白可抑制ccdB大腸桿菌致死基因的表達。植物CRISPR/sgRNA載體[28]pYLgRNAAtU3d-LacZ和 pYLgRNA-AtU3b,為植物CRISPR/Cas9基因編輯載體的中間載體,分別提供small nuclear(sn)RNA U3d和U3b啟動子序列,驅動sgRNA的表達并保證轉錄出的sgRNA停留在細胞核中與Cas9結合,且二者均提供長度為57 nt的sgRNA3′區域保守結構序列,可與靶基因的 20 nt靶序列組裝成sgRNA。此外,pYLgRNA-AtU3d-LacZ還提供了 LacZ標記基因(198 bp)的表達元件,可在含有X-gal的培養基產生藍色菌斑篩選陽性克隆。以上質粒均由華南農業大學劉耀光教授實驗室惠贈。
本文所需引物序列如表1所示,引物合成與序列測序均由上海生工生物工程技術服務公司完成。發根農桿菌 GV3101和大腸桿菌DH5α為本實驗室保存。PCR純化試劑盒與質粒提取試劑盒菌購自全式基因生物有限公司,KOD-Plus酶購自東洋紡(上海)生物科技有限公司,BsaI-HF酶和 T4 DNA連接酶購自New England Biolabs有限公司,其余分子生物學試劑和試劑盒均購自上海生工生物工程技術服務公司。

表1 引物序列Table 1 The list of primer sequences
1.2sgRNA序列設計及分析
為提高打靶效率針對TCS基因設計2個靶點,設計靶點遵循如下原則:目的基因的正鏈和負鏈靶點的打靶效率大致相同,都可以設計靶點;靶點的GC%盡量不要低于40%,靶點序列GC%偏高(50%~70%)有較高的打靶效率;靶點內(按5-N20NGG-3方向)不要有連續4個以上的T,以防RNA Pol III將其作為轉錄終止信號;雖然非特異打靶(脫靶)對植物基因打靶不是很重要的問題,但應進行靶點特異性分析,用靶點+NGG(前后各加幾十堿基)與茶樹轉錄組做 blast分析(參數選擇Somewhat similar sequences),避免使用與同源序列差異少于5個堿基的靶點(在切點附近和PAM有2個堿基差異就具有特異性)。把靶點序列連接到 sgRNA序列的 5′端(target sequence + GTTTTAGAGCTAGAAATAGCA AGTTAAAATAAGGCTAGTCCGTTATCAAC TTGAAAAAGTGGCACCGAGTCGGTGCTTT TTTT),利用在線軟件 RNA Folding Form(http://mfold.rna.albany.edu/?q=mfold/RNA-F olding-Form2.3)做二級結構分析。
1.3sgRNA表達盒構建方法
1.3.1Target 1 sgRNA(T1sgRNA)構建
首先進行第一輪PCR,第一輪PCR進行2個反應。取2~5 ng pYLgRNA-AtU3d-LacZ質粒為模板,在1個PCR反應中使用引物 U-F 和TCSU3dT1;第2個PCR反應中使用引物TCSgRT1和gR-R。第1個PCR和第2個PCR反應分別在兩個 PCR管中進行,采用相同的PCR反應體系(表2)和相同的PCR反應程序:94℃預變性 2 min,25~28循環,94℃ 10 s,58℃ 15 s,68℃ 20 s。取 3 μL反應液進行電泳,檢查產物長度是否符合預期,并估計樣品的大致濃度。第一輪PCR反應完成后,進行第二輪PCR。取第一輪PCR產物U3dT1-gRNA 各1 μL用H2O稀釋10倍,取1 μL為模板,以 Pps-R-C和 Pgs-GG2為引物,擴增相應的U3dT1-gRNA,采用表3所示的反應體系。PCR反應程序:94℃預變性2 min,17~20個循環,94℃ 10 s,58℃ 15s,68℃ 20 s。取 3 μL反應液進行電泳,檢查產物長度是否符合預期,并估計樣品的大致濃度。
1.3.2Target 2 sgRNA(T2sgRNA)構建
首先進行第一輪 PCR,同樣進行 2個反應。取2~5 ng pYLgRNA-AtU3b質粒為模板,在第 1個 PCR反應中使用引物 U-F和TCSU3bT2,第 2個 PCR反應使用引物TCSgRT2和gR-R。U-F/TCSU3bT2擴出U3bT2序列,gR-R/TCSgRT2擴出T2sgRNA序列。采用與構建T1sgRNA第一輪PCR反應相同的PCR反應體系和PCR反應程序。取3 μL反應液進行電泳,檢查產物長度是否符合預期,并估計樣品的大致濃度。第一輪PCR完畢,進行第二輪PCR。取第一輪PCR產物U3bT2-gRNA各1 μL用H2O稀釋10倍,取1 μL為模板,以Pps-GG2和Pgs-L-C為引物,擴增相應的 U3bT2-gRNA。采用與構建T1sgRNA第二輪PCR反應相同的PCR反應體系和PCR反應程序。取3 μL反應液進行電泳,檢查產物長度是否符合預期。
1.4組裝sgRNA表達盒到pYLCRISPR/Cas9載體
1.4.1雙元載體與 sgRNA表達盒的酶切-連接反應
采用PCR產物純化試劑盒對T1sgRNA和T2sgRNA PCR產物純化,使用表4所示的酶切-連接反應體系,反應程序:37℃,1 min,16℃,1 min,30個循環;最后55℃滅活內切酶 5 min。以PCR產物純化試劑盒純化酶切-連接產物。
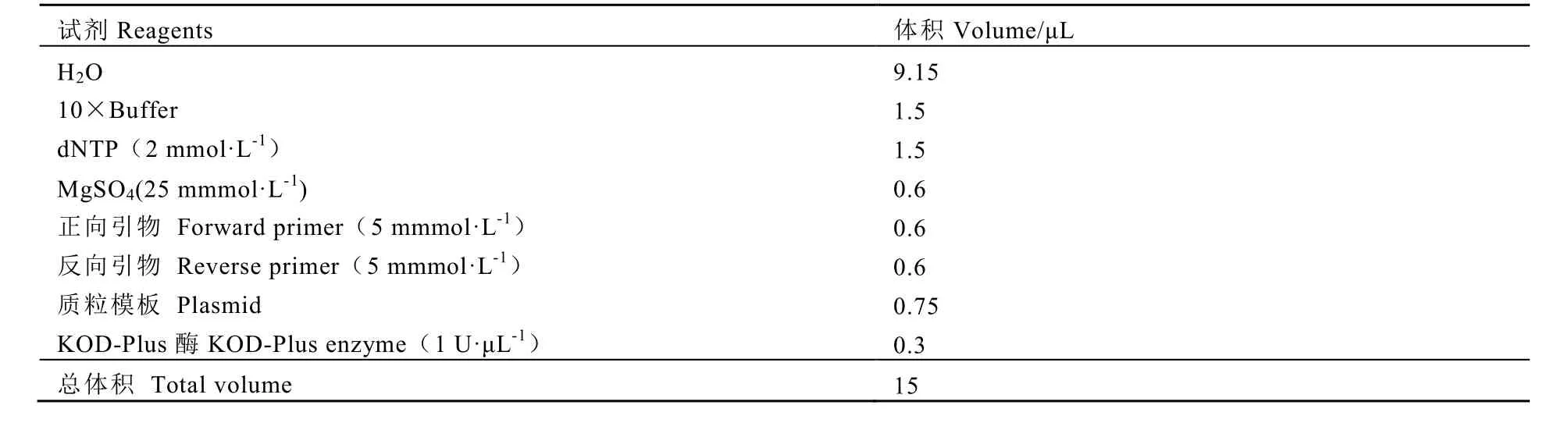
表2 第一輪PCR反應體系Table 2 The PCR reaction system for the first round of PCR

表3 第二輪PCR反應體系Table 3 The PCR reaction system for the second round of PCR
1.5酶切-連接產物的轉化與質粒提取
從-80℃冰箱中取出50 μL DH 5α感受態細胞,放置在冰上直至融化,取10 μL連接產物加入DH 5α感受態細胞離心管,輕輕小彈離心管,使之混合均勻,冰浴20 min,在精確的 42℃水浴中熱擊 90 s,迅速將管子移到冰中放置2 min,加入平衡至室溫的750 μL LB培養基,37℃振蕩培養45 min,取100 μL菌液涂到含50 μg·mL-1Kan、0.5 mmol·L-1IPTG 和40 μg·mL-1X-gal的LB平板,將平板于37℃倒置過夜培養,挑取藍斑菌落,于3 mL含50 μg·mL-1Kan的LB液體培養基中震蕩培養過夜,使用質粒提取試劑盒提取質粒,使用Mlu I酶切驗證,挑取正確質粒進行測序驗證。
2 結果與分析
2.1sgRNA設計
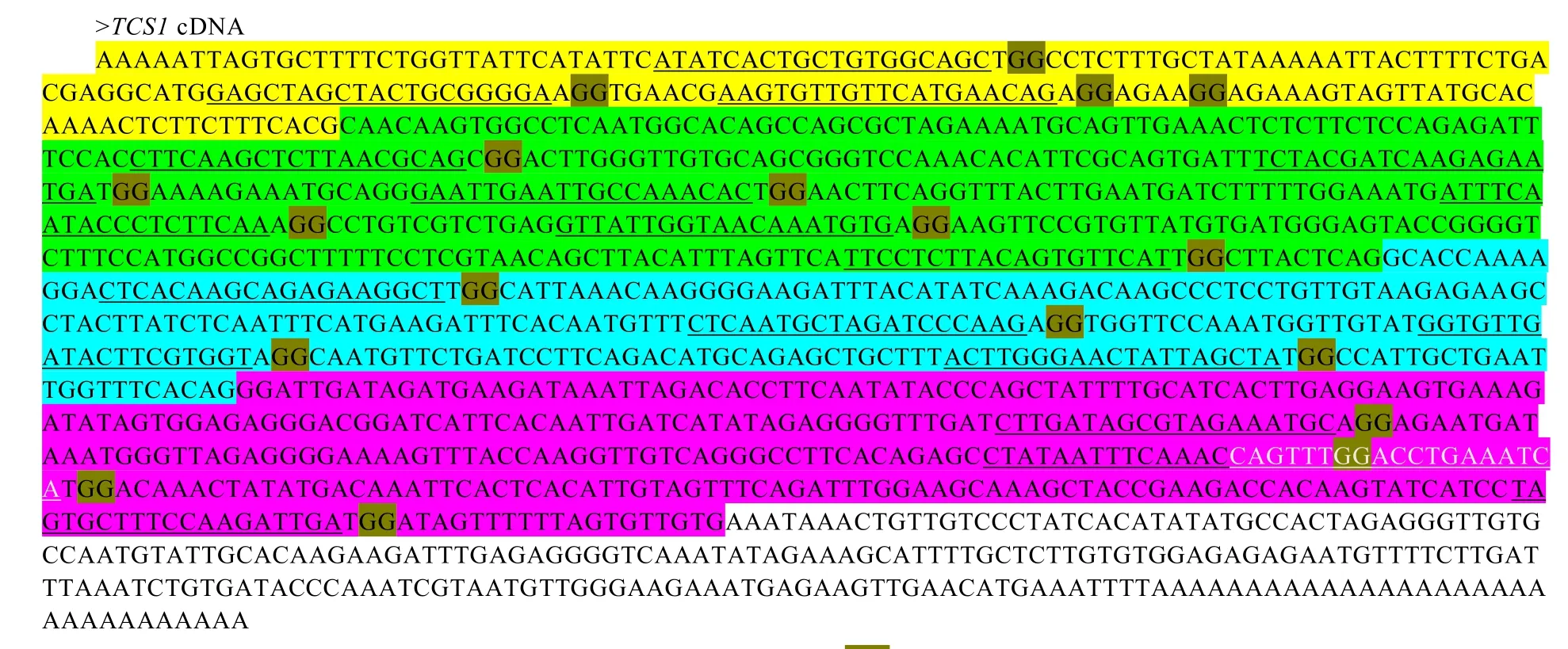
sgRNA序列全長 97 nt,分為兩部分,5′決定靶序列的19 nt種子序列(Seed sequence)和3′區域為保守的結構序列。因此構建針對特定靶位點的 sgRNA,只需要克隆決定靶序列的5′端20 nt。從GenBank中下載茶樹TCS基因組序列和cDNA序列,分析其內含子所在區域。結果表明(圖 2),TCS1基因含 3個內含子。在此基礎上搜尋 TCS的 CRISPR/Cas9靶序列,結果共搜尋到18個不含內含子的候選靶序列。利用RNA Folding Form對18個候選靶序列進行結構分析,靶序列與sgRNA序列產生連續配對 7 bp以上會抑制其與染色體DNA靶序列結合靶點,因此要避免使用連續配對7 bp以上的靶序列。分析結果表明,在TCS1基因 ORF內的靶序列“CTCACAAGCAGA GAAGGCT”(設為靶序列T1)和非編碼區內靶序列“ATATCACTGCTGTGGCAGC”(設為靶序列T2)較為理想,可作為sgRNA序列的種子序列。

表4 雙元載體與sgRNA表達盒的酶切-連接反應體系Table 4 The reaction system for the digestion and ligation of sgRNA expression cassettes and pYLCRISPR/Cas9 vector
2.2質粒提取
將分別含有 pYLgRNA-AtU3d-LacZ、pYLgRNA-AtU3b和pYLCRISPR/Cas9P35S-H質粒的菌劃線活化,挑取單菌搖菌,采用質粒提取試劑盒(小提)提取質粒。電泳結果表明(圖3),提取的質粒符合下一步實驗的要求。
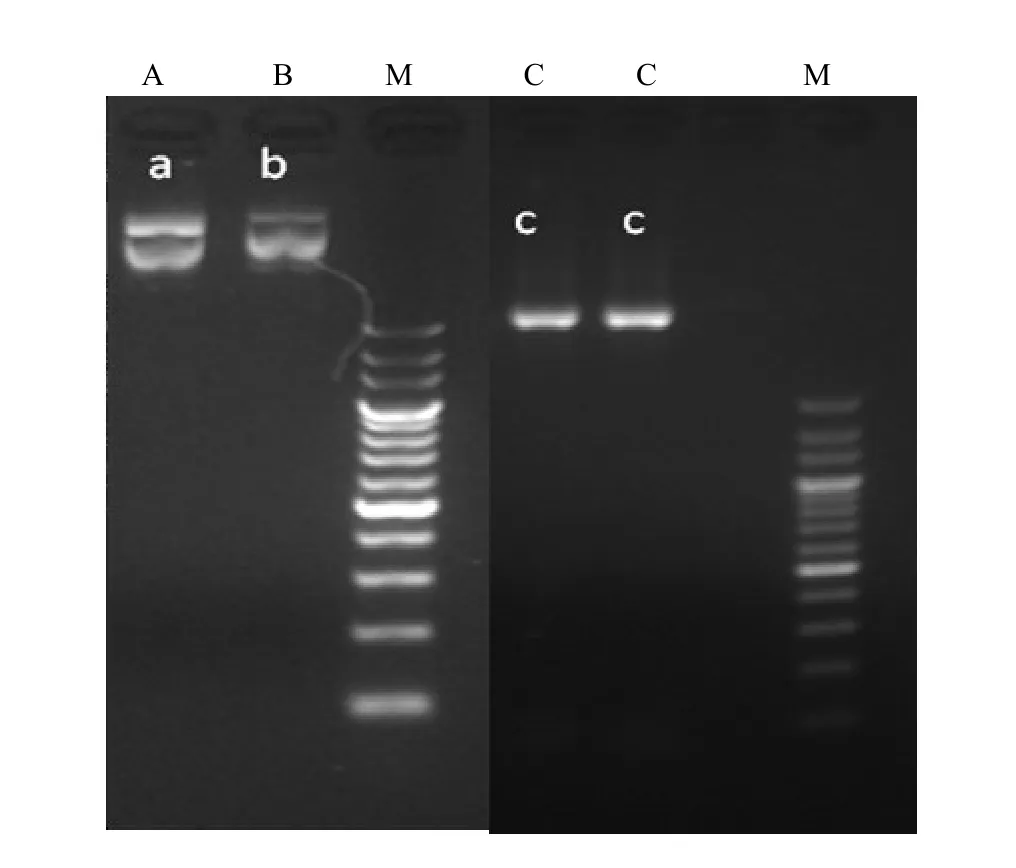
注:A:pYLgRNA-AtU3d-LacZ,B:pYLgRNA-AtU3b,C:pYLCRISPR/Cas9P35S-H,M:marker。Note:A:pYLgRNA-AtU3d-LacZ,B:pYLgRNA-AtU3b,C:pYLCRISPR/Cas9P35S-H,M:marker.圖3 質粒電泳圖Fig.3 The electrophoresis analysis of plasmids
2.3sgRNA表達盒的構建
sgRNA需要在植物細胞內合成從而指導Cas9蛋白編輯靶基因,而sgRNA的合成是通過sgRNA表達盒來完成,sgRNA表達盒由啟動子序列和gRNA序列組成。pYLgRNA載體(pYLgRNA-AtU3d-LacZ、pYLgRNA-AtU3b)提供了不同的snRNA啟動子可在植物細胞核內轉錄gRNA,并使gRNA停留在細胞核內發揮其功能。此外,pYLgRNA載體還提供gRNA中保守序列。因此,以pYLgRNA質粒為模板,通過常規PCR可以獲得snRNA啟動子序列和gRNA中保守序列,而gRNA中5′段的種子序列在引物合成時導入,聯合應用常規PCR和overlapping PCR將啟動子序列和gRNA序列組裝在一起(圖4)。電泳分析表明(圖5),針對靶序列 T1的第一輪PCR獲得了 360 bp 和130 bp左右的兩個片段,分別為U3d啟動子和連接T1靶序列的gRNA;而針對靶序列T2的第一輪PCR獲得了380 bp和130 bp左右的兩個片段,分別為U3b啟動子和連接T1靶序列的gRNA;針對靶序列T1的第二輪PCR 將U3d啟動子片段和gRNA片段2個分離的片段連接在一起組裝成T1sgRNA表達盒;針對靶序列T2的第二輪PCR將U3b啟動子片段和gRNA片段2個分離的片段連接在一起組裝成T2sgRNA表達盒。
2.4兩個sgRNA表達盒的拼接及與Cas9雙元表達載體的組合
T1sgRNA和T2sgRNA兩個單獨的表達盒構建好后,需要將兩個表達盒拼接在一起,并且插入Cas9雙元表達載體中,以便通過農桿菌的介導整合到茶樹基因組中,從而在茶樹細胞內進行表達,獲得Cas9和gRNA,進而對目標基因進行編輯。我們應用 Golden gate cloning 技術,一步完成sgRNA表達盒的拼接及與 Cas9雙元表達載體的組合,從而獲得CRISPR/Cas編輯載體(圖6)。如圖7所示,酶切、連接產物可見一條 980 bp左右的電泳條帶,表明T1sgRNA和T2sgRNA表達盒連接成功,同時還可見一條>4 000 bp的電泳條帶,說明T1sgRNA和T2sgRNA表達盒可能已組裝進入 pYLCRISPR/Cas9P35S-H雙元表達載體。連接產物轉化 DH5α后搖菌提取質粒,以Mlu I酶切,結果如圖所示,可見兩條電泳條帶,其中一條 950 bp左右的條帶為T1sgRNA和T2sgRNA連接片段,而pYLCRISPR/Cas9P35S-H雙元表達載體未見950 bp左右的電泳條帶,說明酶切、連接成功,獲得了TCS CRISPR/Cas9基因編輯載體。
2.5測序驗證
為了進一步驗證TCS 的CRISPR/Cas9基因編輯載體是否構建成功,將獲得的10個質粒送上海生物工程技術有限公司測序,測序引物使用 SP-L2和 SP-R-C。測序結果(圖 8)表明,T1sgRNA和T2sgRNA表達盒構建成功,在119 bp處檢測到T1、T2靶序列,緊接靶序列后檢測到 gRNA保守序列的存在,同時在T1和T2靶序列上游分別檢測到U3d和U3b啟動子序列,在 T1sgRNA 表達盒上游T2sgRNA表達盒下游均檢測到了pYLCRISPR/Cas9P35S-H雙元表達載體序列。測序結果(圖 9)說明,T1sgRNA表達盒和T2sgRNA 表達盒構建成功,并成功組裝入pYLCRISPR/Cas9P35S-H雙元表達載體,證實TCS的CRISPR/Cas9基因編輯載體構建成功。
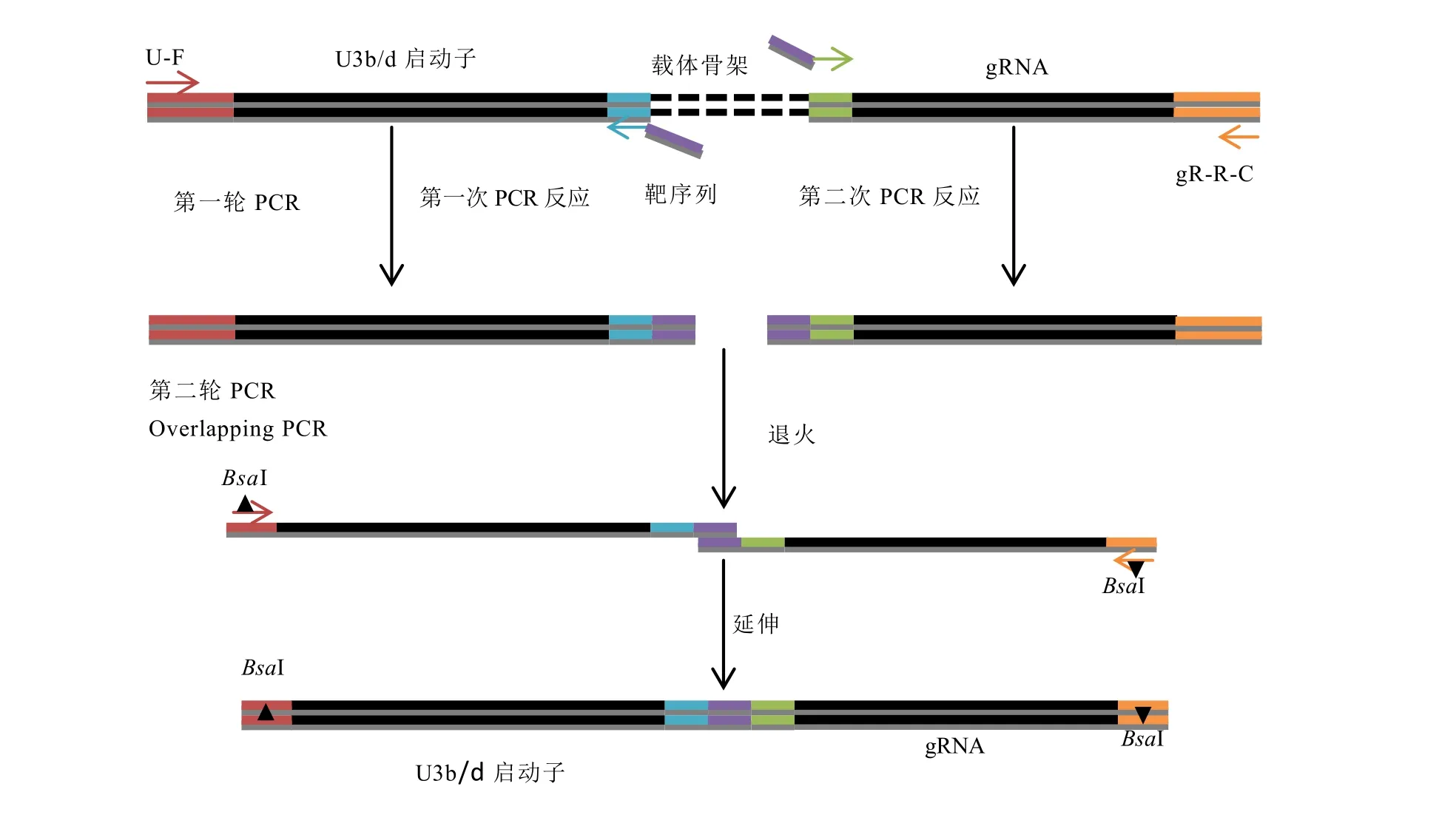
圖4 sgRNA表達盒的構建流程Fig.4 Procedures for generation of a sgRNA expression cassette
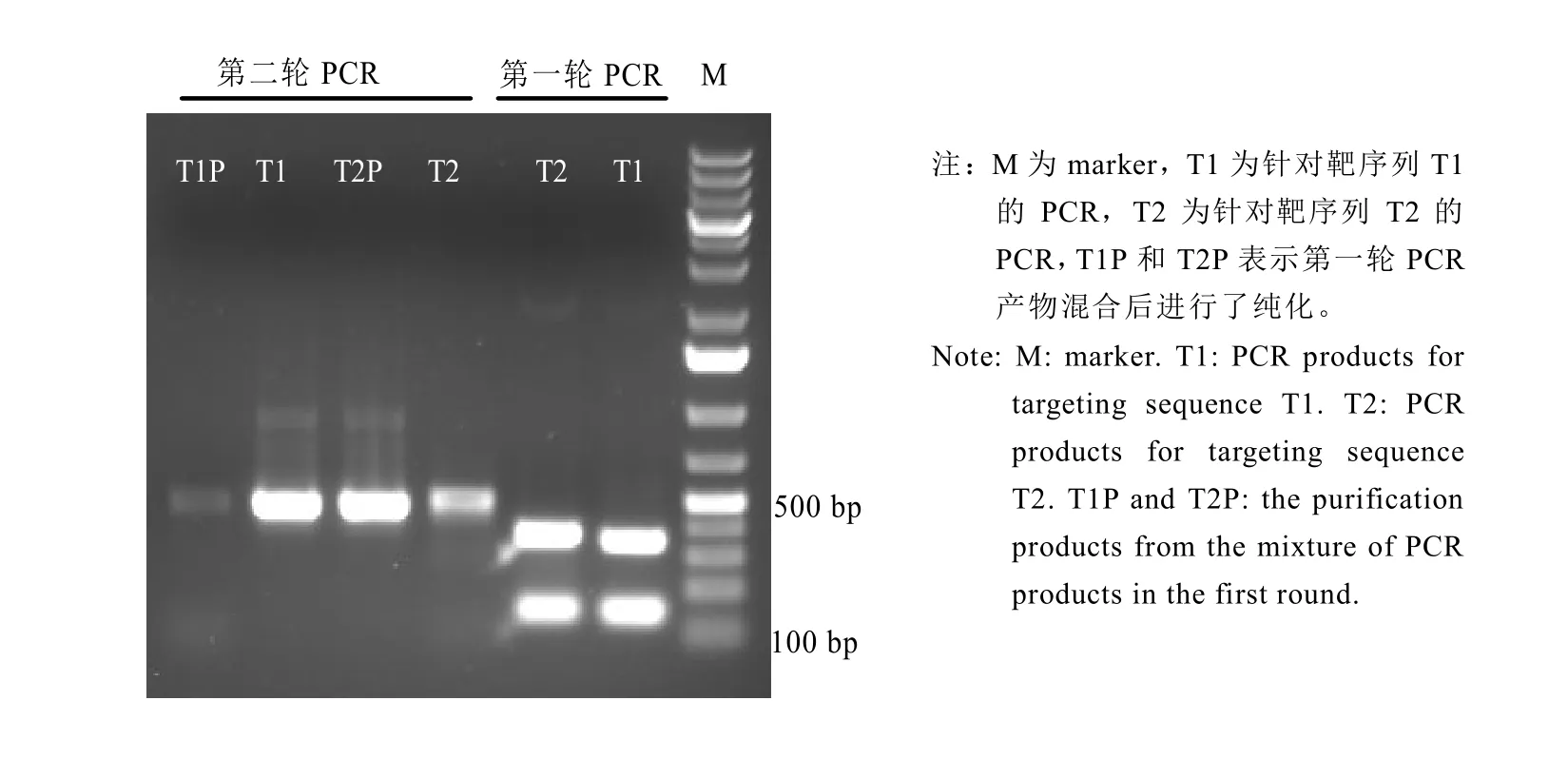
圖5 第一、二輪PCR反應產物電泳圖Fig.5 The electrophoresis analysis of the first and the second round of PCR products
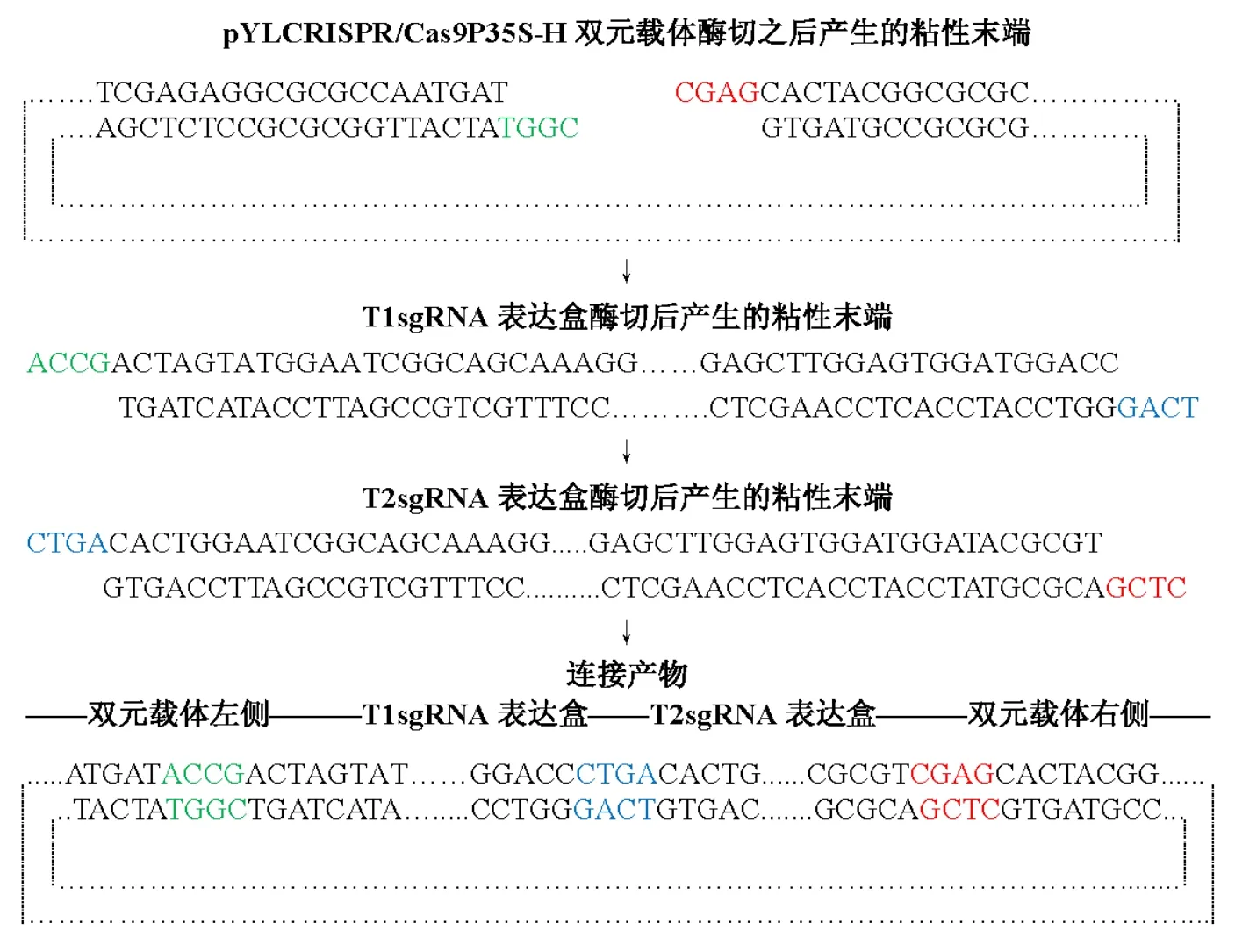
圖6 Golden gate cloning 組裝茶樹CRISPR/Cas9基因組編輯載體Fig.6 Asembly the CRISPR/Cas9 editing vector by using golden gate cloning.
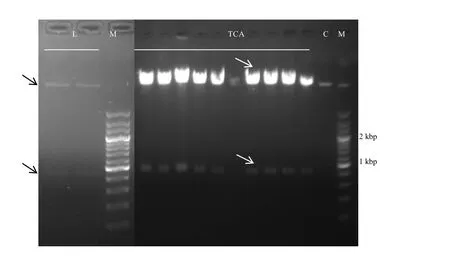
注:M為maker;L為sgRNA表達盒與Cas9雙元表達載體酶切、連接產物;TCA為CRISPR/Cas9基因編輯載體的Mlu I酶切產物;C為質粒pYLCRISPR/Cas9P35S-H的Mlu I酶切產物。Note:M:maker.L:Digestion and ligation products of sgRNAs and pYLCRISPR/Cas9P35S-H.TCA:Digestion products of CRISPR/Cas9 cassettes for TCA with Mlu I.C:Digestion products of pYLCRISPR/Cas9P35S-H with Mlu I.圖7 連接產物及質粒酶切電泳圖Fig.7 The electrophoresis analysis of ligation and digestion products
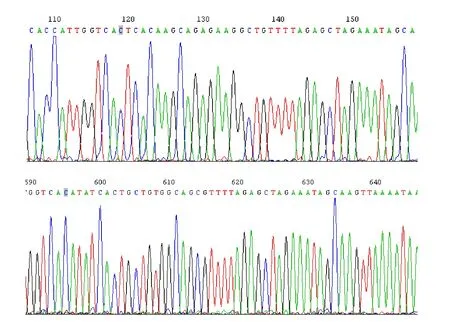
圖8 TCS基因編輯載體部分測序圖Fig.8 The sequencing chromatogram of genome editing vector for TCS

圖9 TCS基因編輯載體測序結果Fig.9 The sequence of genome editing vector for TCS
3 討論
CRISPR是一種簡單的,在全基因組水平上選擇性調控目的基因表達的方法。該技術不同于傳統的RNA干擾技術(RNAi),CRISPR干擾可以同時沉默任意數量的單個基因。此外,它能夠更明確、更穩定地發揮作用,能夠極其靈活和快速選擇靶位點。2013年,張峰等[14]首先利用 CRISPR/Cas9技術實現了真核生物基因組的剪切。此后,CRISPR/Cas9技術研究一發不可收拾,國內外學者迅速在多種動、植物中開展了CRISPR/Cas9研究,不到3年的時間就已經在模式植物、農作物及園藝植物中取得了非常成功的應用[17]。據報道,在擬南芥和水稻中,CRISPR/Cas9系統引起的突變率高達90%以上。通常情況下,CRISPR/Cas9會產生野生株、雜合突變體、雙等位基因突變體和純合突變體,而只有雙等位基因突變體和純合突變體才有應用價值。因此,如果第一代轉基因未能獲得雙等位基因突變體或純合突變體需要通過不斷自交從子代中篩選雙等位基因突變體和純合突變體。幸運的是,業已證明多種植物均能在第一代轉基因中獲得雙等位基因突變體或純合突變體,從而增加了 CRISPR/Cas9實用性。因此,對于茶樹等自交不親和的作物同樣可以采用CRISPR/Cas9技術獲得純合突變株。但是,將CRISPR/Cas9技術應用于茶樹中將面臨表達載體構建的問題,目前尚無相關報道。本文以茶樹中咖啡堿合成酶為例,聯合采用常規PCR、Overlapping PCR和Golden Gate Cloning技術,構建了包含茶樹咖啡堿合成酶雙靶點的CRISPR/Cas9基因編輯載體,為CRISPR/Cas9基因編輯技術在茶樹中的應用奠定了理論基礎,同時對其他植物 CRISPR/Cas9基因編輯載體的構建提供了技術支撐。
雖然茶樹基因組尚未釋放,影響脫靶效應分析。但是,一般來說 CRISPR/Cas9要對基因編碼區進行編輯才能引起突變表型,在沒有全基因組序列的情況下對轉錄組序列進行比對,可在一定程度上排除錯靶現象。此外,相對動物而言,植物的脫靶帶來的風險較小。所以,在茶樹基因組釋放前,同樣可以開展針對茶樹的CRISPR/Cas9研究工作。
本文建立的 CRISPR/Cas9基因組編輯載體的構建方法,在Ma等[28]報道的方法上進行了完善與改進。pYLCRISPR/Cas9P35S-H等雙元表達載體因屬于低拷貝載體,且載體較大,一般需要采用大提質粒試劑盒來提取,具有成本高、效率低的缺點。本文通過擴大菌的培養量和培養時間,菌液約5 mL左右,震蕩培養約 16 h,洗脫質粒時使用 60℃無菌水,且在烘箱中60℃孵育2 min,最后發現,采用小提質粒提取試劑盒同樣可以獲得高質量且足量的pYLCRISPR/Cas9P35S-H質粒(圖3),完全滿足載體構建要求。
第一輪PCR的目的是將gRNA及其啟動子分別克隆出來,而第二輪 PCR是利用Overlapping PCR將和啟動子連接起來形成gRNA表達盒,并在gRNA表達盒兩端加上帶BsaI酶切位點的接頭,然后酶切連接,將各個gRNA表達盒連接起來,并與pYLCRISPRcas9載體連接。在第一輪PCRMa等[28]人將第一個反應與第2個反應在同一PCR管中進行,在一個反應中使用4種引物,在循環初始階段同時擴增兩個片段,并在循環后階段通過Overlapping PCR將兩片段拼接在一起。然而,我們實驗結果表明,同一PCR管中進行2個反應很難獲得特異片斷。最后,我們將2個反應分開進行,獲得了理想的結果。
本載體系統采用 Golden Gate Cloning[29]技術組裝Cas9載體和多個gRNA表達盒片段。Golden Gate cloning是利用 BsaI(type IIs restriction enzyme)的識別位點和切割位點不重疊的特性,可以設計出多種不同的且非回文結構的粘性末端(256-16=240種,減去的 16種是回文結構)。多個要連接的片段在連接點具有特異的互補末端可以連接,而非連接點的末端之間不互補不能連接(相同末端分子間也不互補不能連接),因此連接反應是向著目標單方向進行,效率很高。此外,利用 BsaI酶進行Golden Gate cloning時,可實現酶切、連接同步化,即邊切邊連,不需要酶切后切膠回收的繁瑣操作,簡單、方便且節省大量時間。Ma等[28]采用的酶切、連接程序為:10℃ 5 min,20℃ 5 min;10~15循環。按照Ma等[28]等方法我們嘗試多次,但均未成功,后來對反應條件進行了多次優化,最后確定了酶切、連接程序為:37℃ 1 min,16℃ 1 min,30個循環,最后55℃滅活內切酶5 min。
本文成功構建了靶向TCS的CRISPR/Cas9基因組編輯載體,為茶樹其他基因靶向的 CRISPR/Cas9基因組編輯載體的構建提供了借鑒方法。下一步的工作,是通過根癌農桿菌的介導將 CRISPR/Cas9基因組編輯載體轉入茶葉愈傷組織,獲得轉基因愈傷組織或轉基因茶樹,并檢測突變材料中的咖啡堿生物合成情況,驗證 CRISPR/Cas9基因組編輯載體的有效性。如有必要,再對載體進行改進,比如使用茶樹本身的 sn RNA啟動子驅動gRNA的表達,根據茶樹的特性優化 Cas9密碼子,從而建立完善的 CRISPR/Cas9介導的茶樹基因組編輯體系,推動茶樹分子育種、基因功能研究、茶樹基因工程等工作快速發展。
[1]Shi CY,Yang H,Wei CL,et al.Deep sequencing of the Camellia sinensis transcriptome revealed candidate genes for major metabolic pathways of tea-specific compounds [J].BMC Genomics,2011,12(1):1-19.
[2]Liu SC,Jin JQ,Ma JQ,et al.Transcriptomic analysis of tea plant responding to drought stress and recovery [J].PLoS ONE,2016,11(1):e0147306.http://dx.doi.org/10.1371/journal.pone.0147306.
[3]Wei Y,Jing W,Youxiang Z,et al.Genome-wide identification of genes probably relevant to the uniqueness of tea plant(Camellia sinensis)and its cultivars [J].International Journal of Genomics,2015,2015:527054.doi:10.1155/2015/527054.
[4]Wang XC,Zhao QY,Ma CL,et al.Global transcriptome profiles of Camellia sinensis during cold acclimation [J].BMC Genomics 2013,14:415.DOI:10.1186/1471-2164-14-415.
[5]Mukhopadhyay M,Mondal TK,Chand PK.Biotechnological advances in tea(Camellia sinensis [L.]O.Kuntze):a review [J].Plant Cell Reports,2015,35(2):255-287.
[6]Fauser F,Roth N,Pacher M,et al.In planta gene targeting [J].Proc Natl Acad Sci USA,2012,109(19):7535-7540.
[7]Li T,Liu B,Spalding MH,et al.High-efficiency TALEN-based gene editing produces disease-resistant rice [J].Nat Biotech,2012,30(5):390-392.
[8]Belhaj K,Chaparro-Garcia A,Kamoun S,et al.Editing plant genomes with CRISPR/Cas9 [J].Current Opinion in Biotechnology,2015,32:76-84.
[9]Mali P,Yang LH,Esvelt KM,et al.RNA-Guided human genome engineering via Cas9 [J].Science,2013,339(6121):823-826.
[10]Xie K,Yang Y.RNA-guided genome editing in plants using a CRISPR-Cas system [J].Mol Plant,2013,6(6):1975-1983.[11]Schaeffer SM,Nakata PA.CRISPR/Cas9-mediated genome editing and gene replacement in plants:transitioning from lab to field [J].Plant Sci,2015,240:130-142.
[12]Wiedenheft B,Sternberg SH,Doudna JA.RNA-guided genetic silencing systems in bacteria and archaea [J].Nature,2012,482(7385):331-338.
[13]Jinek M,Chylinski K,Fonfara I,et al.A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity [J].Science,2012,337(6096):816-821.
[14]Wang HY,Yang H,Shivalila CS,et al.One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering [J].Cell,2013,153(4):910-918.
[15]Schiml S,Puchta H.Revolutionizing plant biology:multiple ways of genome engineering by CRISPR/Cas [J].Plant Methods,2016,12(1):1-9.
[16]Lintner NG,Frankel KA,Tsutakawa SE,et al.The structure of the crispr-associated protein csa3 provides insight into the regulation of the CRISPR/Cas system [J].Journal of Molecular Biology,2011,405(4):939-955.
[17]Gilbert LA,Larson MH,Morsut L,et al.CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes [J].Cell,2013,154(2):442-451.
[18]Nekrasov V,Staskawicz B,Weigel D,et al.Targeted mutagenesis in the model plant Nicotiana benthamiana using Cas9 RNA-guided endonuclease [J].Nat Biotech,2013,31(8):691-693.
[19]Jiang WZ,Zhou HB,Bi HH,et al.Demonstration of CRISPR/Cas9/sgRNA-mediated targeted gene modification in Arabidopsis,tobacco,sorghum and rice [J].Nucleic Acids Res,2013,41(20):e188.
[20]Wang Z,Xing H,Dong L,et al.Egg cell-specific promoter-controlled CRISPR/Cas9 efficiently generateshomozygous mutants for multiple target genes in Arabidopsis in a single generation [J].Genome Biol,2015,16:144.
[21]Upadhyay SJ,Alok A,Tuli R.RNA-guided genome editing for target gene mutations in wheat [J].G3(Bethesda).2013,3(12):2233-2238.DOI:10.1534/g3.113.008847.
[22]Liang Z,Zhang K,Chen KL,et al.Targeted mutagenesis in Zea mays using TALENs and the CRISPR/Cas system [J].J Genetics Genomics,2014,41(2):63-68.
[23]Zhang H,Zhang J,Wei P,et al.The CRISPR/Cas9 system produces specific and homozygous targeted gene editing in rice in one generation [J].Plant Biotechnol J,2014,12(6):797-807.
[24]?ermák T,Baltes NJ,?egan R,et al.High-frequency,precise modification of the tomato genome [J].Genome Biol,
[25]Jia H,Wang N.Targeted genome editing of sweet orange using Cas9/sgRNA [J].PLoS ONE,2014,9(4):e93806.DOI:10.1371/journal.pone.0093806.
[26]Fan D,Liu T,Li C,et al.Efficient CRISPR/Cas9-mediated targeted mutagenesis in populus in the first generation [J].Scientific Reports,2015,5:12217.
[27]Zhang B,Yang X,Yang C,et al.Exploiting the CRISPR/Cas9 system for targeted genome mutagenesis in petunia [J].Scientific Reports,2016,6:20315.
[28]Ma X,Zhang Q,Zhu Q,et al.A Robust CRISPR/Cas9 system for convenient,high-efficiency multiplex genome editing in monocot and dicot plants [J].Mol Plant,2015,8(8):1274-1284.
[29]Marillonnet S,Werner S.Assembly of multigene constructs using golden gate cloning [J].Methods Mol Biol,2015,1321:269-284.
Development of a CRISPR/Cas9 Constructed for Genome Editing of Caffeine Synthase in Camellia sinensis
TANG Yuwei1,3,LIU Liping1,3,WANG Ruoxian1,CHEN Yuhong1,LIU Zhonghua1,2,3,LIU Shuoqian1,2,3*
1.College of Horticulture and Hardening,Hunan Agricultural University,Changsha 410128,China;
2.National Research Center of Engineering Technology for Utilization of Functional Ingredients from Botanicals,Changsha 410128,China;
3.Key Lab of Tea Science,Ministry of Education,Changsha 410128,China
CRISPR/Cas9 technology(clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9)is a novel and powerful approach for targeted genome editing,such as targeted gene knock out or site-directed mutagenesis in a simple and easy way.Since its establishment,the CRISPR/Cas9 technique has been successfully applied in many eukaryotic organisms,including more than 10 plant species.However,it has not been available for genome editing of tea plant [Camellia sinensis(L.)O.Kuntze]due to the difficulty in constructing CRISPR/Cas9 expression vector.The present work developed an efficient method to construct a CRISPR/Cas9 expression vector for genome editing a tea caffeine synthase(TCS)by using general PCR,overlapping PCR and golden gate cloning technology.The present work would promote the application of CRISPR/Cas9 technology in genomic modification in tea plants.
Camellia sinensis(L.),genome editing technology,tea caffeine synthase,CRISPR/Cas9 technique
TS272;Q52
A
1000-369X(2016)04-414-13
2016-04-25
2016-05-12
唐雨薇,女,碩士研究生,主要從事茶樹分子生物學研究,*通訊作者:shqianliu@sina.com
2015,16:232.10.1186/s13059-015-0796-9.
