基于Map Reduce的多序列星比對方法在腫瘤研究中的應用△
2016-10-18 01:28:38李大鵬鞠穎鄒權
癌癥進展 2016年6期
關鍵詞:方法
李大鵬 鞠穎 鄒權
1秦皇島市第四醫院腫瘤內科,河北秦皇島066000
2廈門大學計算機科學系,福建廈門361005
3天津大學計算機科學與技術學院,天津300072
基于Map Reduce的多序列星比對方法在腫瘤研究中的應用△
李大鵬1鞠穎2鄒權3#
1秦皇島市第四醫院腫瘤內科,河北秦皇島066000
2廈門大學計算機科學系,福建廈門361005
3天津大學計算機科學與技術學院,天津300072
序列比對是生物信息學的基礎,通過多條序列比對可以挖掘出生物序列中的各種重要信息。大規模的基因序列比對方法對運算能力要求較高,基于Map Reduce框架的多序列比對方法在多序列星比對算法的基礎上利用分布式并行計算來處理大規模數據。實驗結果表明:相對于單機處理方法,基于Map Reduce的序列比對方法可以更快速地處理大規模數據,并且具有良好的硬件擴展性。本文探討了多序列比對在腫瘤研究方面的應用前景。
生物信息學;多序列比對;星比對算法;Map Reduce;并行計算;癌癥
序列比對是通過對基因序列的比較找出序列之間的相似性和同源性,以便通過已知序列的結構和功能信息來預測未知基因序列所具有的生物學信息。雙序列比對通過添加空格或者刪除字母使得給定的兩條序列具有最大的相似性。多序列比對同時比對多條序列,使得這組序列之間具有最大的相似性。目前已有許多序列比對軟件被開發出來,如Clustal[1]、T-coffee[2]、MAFFT[3]和HMMER[4]等。但是隨著序列數目的急劇增加,海量的數據對這些軟件的計算成本造成了嚴峻的挑戰,因此開發出一種能處理大規模數據并具有良好擴展性的軟件是一項重要的工作。
在多序列比對算法方面,目前已取得了一定的成果。基于關鍵字樹的DNA多序列星比對算法[5]利用Aho-Corasick搜索算法(簡稱AC自動機)進行模式匹配,以便于選取中心序列,進一步用星比對算法進行序列比對。還有研究人員提出利用啟發式算法解決多序列比對問題,包括基于A*算法的啟發式算法[6],基于模擬退火的多序列比對算法[7],基于遺傳算法的多序列比對算法[8],基于粒子群算法的多序列比對算法[9]。也有研究人員利用概率模型進行多序列比對,如基于隱馬爾可夫模型的比對算法[10]。然而以上算法都無法有效處理基因組規模或幾百條序列以上的大數據。
隨著科學技術的不斷發展,生物信息數據,氣象監測數據,社交網絡數據呈爆炸性增長,快速有效的處理海量數據已成為急需解決的問題。Google公司提出了Map Reduce并行計算框架用于處理大規模的并行計算問題。Apache基金會基于Map Reduce框架開發出了java編程平臺Hadoop[11],可以實現多機并行處理大規模數據。本文利用Map Reduce框架,結合基于k-band的多序列星比對算法[12-13],引入并行計算來處理大規模數據,實驗結果表明:相對于單機處理程序,本文提出的方法能顯著提高多序列比對的運算效率。
1 基于Map Reduce框架的多序列比對方法
1.1空隙罰分
由于序列比對是求解序列之間最大相似性的問題,所以需要一個判斷相似性的度量方法,于是引入了空隙罰分機制。對兩個或者多個字符串序列通過匹配相應的字符或者插入空格從而得到序列之間的最大相似性。雖然加入空格匹配序列的方法能夠獲得較好的匹配效果,但是極可能會破壞其生物學意義。空隙罰分就是用來防止無限制地引入空格,補償插入和缺失對序列相似性的影響。對于一個空格數為p的罰分計算公式如下:

其中a表示空位罰分(gap opening),即每插入一個空隙的罰分值,b表示空位擴展罰分(gap extension),即每添加一個空格的罰分值,p為插入的空格數。
對于多序列比對假設有基因序列s={s1,s2,……sn},用score(x,y)表示兩條序列x和y之間的比對得分,那么這組基因序列的比對分值spscore(s)就表示為:

1.2多序列星比對算法
動態規劃算法是最常用于雙序列比對的算法,即Needleman和Wunsch提出的Neeedlemana-Wunsch算法[14]。由于動態規劃的計算時間復雜度為O(n^2),但是待比對的序列差異性通常不會太大,動態規劃的主要回溯路徑集中在二維表的主對角線上,為減小算法的時間復雜度以及空間復雜度,在計算動態規劃比對算法的時候可以以主對角線為中心,取寬度為k的一條窄帶上的相似性分值。對于兩條長度分度分別為n,m的不等長序列,假設m>n,k的寬度初始時至少為m-n,此時最差的相似性分值為M(m-k-1)-2(k+1)(a+b),其中M為字符匹配的得分值,a為空隙罰分,b為空格罰分,如果計算出的相似性分值小于最差的相似性分值,就需要適當地擴大k的數值,從而找到更好的序列比對方案。
星比對算法就是挑選一條序列作為中心序列,用這條中心序列和其他序列依次進行比對,從而獲得比對后的更新序列以及新的中心序列。對于相似性較高的基因序列,先任意選取一條序列作為中心序列,用該中心序列依次與其他序列相互比對,然后匯總更新后的中心序列,獲得最終比對后的中心序列,再利用這條中心序列對第一次更新的序列進行二次修正,獲得最終的更新序列組,具體計算方法如下。
星比對算法:

1.3Map Reduce的實現方法
由于Map Reduce每條記錄是以(key,value)形式輸入,其中key用序列名表示,value用基因序列所代表的堿基字符來表示。對于用戶輸入的文件可能會存在錯誤,如文件格式不滿足要求,序列字符可能出現非法字符的情況要進行辨別與修正。首先把文件轉化為適應hadoop的鍵值對模型,在轉化文件結束后,提取序列組中的第一條序列并保存,作為中心序列。
然后進入Map Reduce框架進行序列比對計算,在第一個Map函數處理階段,數據文件會被自動分割成一個個大小為64 M的split文件,每個Map函數在不同的Data Node節點機上執行,中心序列同時和每個Map塊上的序列進行比對,每次比對會生成兩條更新序列,一條是原中心序列的更新(New_center_sequence[i]),一條是序列i的更新(New_sequence[i]),然后將比對的結果保存為(sequence_name,value)的格式,其中value的值為New_center_sequence[i]與New_sequence[i],這兩條序列之間用tab分隔符隔開,Map_1函數輸入與輸出結果,詳見圖1。

圖1 第一階段Map函數輸入與輸出

進入reduce階段后,reduce函數并不對數據進行處理,直接把結果輸出到HDFS文件系統下。隨后從HDFS文件系統中將結果復制回本地文件系統,并從文件中分離出更新中心序列(New_center_sequence[i])。隨后對更新中心序列進行匯總,匯總的方法即對于n條更新序列,統計各條序列各個空隙所具有的空格數,求出相應空隙所具有的最大空格數,即為最終中心序列(Final Center Sequence)的空隙插入方式,接著進入第二階段Map Reduce,具體計算方法如下。
中心序列匯總:


第二階段Map Reduce的過程基本和第一階段相同,采用匯總后的中心序列與第一階段輸出的更新序列(New_sequence[i])再次進行比對。不同之處在于,Map函數并不保存比對后的中心序列,只保存二次比對序列i的最終更新結果(Final_new_sequence[i]),Map_2函數輸入與輸出,詳見圖2。
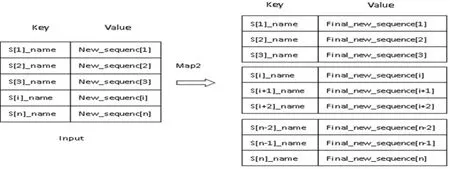
圖2 第二階段Map函數輸入與輸出
2 實驗結果
實驗基于Hadoop1.0版本完成,集群配置分別采用1個Name Node,2個Data Node以及1個Name Node,4個Data Node兩組對比實驗,用于對比硬件擴展對于運算效率的影響。同時添加了一組單機版本的實驗,以觀測并行計算相對于單處理在運算效率上是否有所提高。實驗運行環境配置如下:單機以及集群的Name Node節點均采用64位Ubuntu操作系統,CPU為Intel(R)Core(TM)i7-3820@3.60 GHz,內存8 G*8。集群的Data Node節點采用64位Ubuntu操作系統,CPU為Intel(R)Xeon(R)E31230@3.2 GHz,內存4 G*8。
為便于比對不同數據規模對于運算性能的提升,實驗采用大小分別為1 G、2 G、4 G、8 G的人類Mitochondrial DNA數據(每條序列的長度約為16 kbp)進行分組觀察,統計程序5次運算時間并選取其中位數(圖3),從圖中可以觀測得到,在相同數據規模的情況下,基于Map Reduce框架的多序列比對程序相對于單機程序所需運算時間降低,同時隨著數據規模的增大,程序的運算效率進一步提高,這表明基于Map Reduce框架的多序列比對程序能很好地處理大規模數據。
由于單機處理所需運算時間的基數過大,圖3所示的不同配置集群在運算效率提升上的差異性不是特別明顯,由此引入加速比來衡量并行系統或程序并行化性能。加速比是同一個任務在單處理器系統和并行處理器系統中運行消耗時間的比率,計算公式如下:
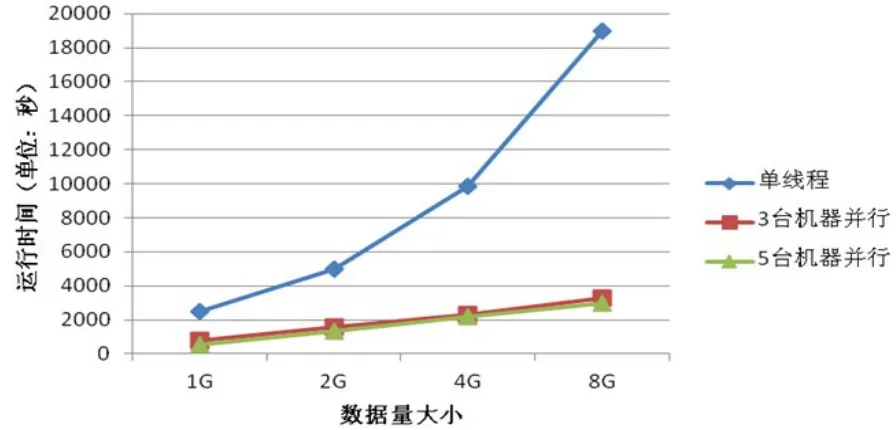
圖3 不同集群配置在不同數據規模上的運算時間

其中Sp為系統加速比,T1是單處理器下的運行時間,Tp是并行系統中的運行時間。統計不同配置集群的加速比結果(圖4),可以直觀的觀測到隨著硬件配置的擴展,系統運算效率的提升表現更加明顯。當數據規模進一步增大時,適當的擴充現有的硬件條件,可以獲取更高的運算效率。
本文提出了一種基于Map Reduce框架,結合星比對算法的多序列比對方法,通過引入并行計算來提升程序的運行效率。實驗證明:隨著問題規模的擴大,與單機單線程的多序列比對軟件相比[1,5],基于Map Reduce的多序列比對方法能提升序列比對的運算效率,同時程序具有良好的硬件擴展能力。為了方便研究使用,作者還開發了可以在Hadoop平臺上使用的多序列比對軟件MSA Hadoop1.0(http://datamining.xmu.edu.cn/software/MSA Hadoop),研究者可以在上述網址中下載到本文實驗中提及的軟件以及操作指南。
3 多序列比對在腫瘤研究中的應用
多序列比對的結果有助于發現單核苷酸多態性(SNP),拷貝數差異(CNV)和序列缺失(indel)。而已有大量研究表明,腫瘤的產生機制,遺傳效應和變異程度均與上述現象相關。2004年,日本研究者通過對人類的線粒體DNA進行測序,然后利用多序列比對的方法發現,阿茲海默癥和糖尿病均與線粒體DNA上的若干敏感位點相關[15]。此外,通過多序列比對和將測序序列向基因組上回貼(mapping),也可以找出腫瘤患者與正常人在部分基因上的表達差異,相關研究發現MicroRNA的表達差異與腫瘤有密切關系[16-17]。還有研究發現,MicroRNA不但在表達量上產生差異,而且成熟體的序列上也有細微差異,這種差異被稱為isomiR(miRNA的多樣性表達)[18],這些isomiR的比較分析依然需要多序列比對的方法。目前在腫瘤遺傳學研究中,盡管對SNP難以定義,但全基因組關聯分析(GWAS)依然是主要的研究方法。從腫瘤細胞到癌旁細胞,從未轉移的腫瘤細胞到轉移的腫瘤細胞的對比分析依然需要依靠GWAS,而相關的測序數據不但要映射回基因組找出差異,還需要在立體的層面對所有的位點進行對齊和分析,隨著GWAS研究的深入,已不僅是對SNP位點進行關聯分析[19],還需要更多的情況考慮序列的插入和缺失,以及各種基因的表達量等,這對多序列比對提出了更高的挑戰。因此,關于多序列比對的理論與內容還要面對更大規模的數據研究,以及融合入更多信息的復雜模型。
[1]ThomposonJD,GibsonTJ,HigginsD.CLUSTALW:improving the sensitivity of progressive multiple sequence alignment through sequence weighting position-specific gap penalties and weight matrix choice[J].Nucleic Acids Res,1994,22(22):4673-4680.
[2]Noredama C,Holm L,Higgins DG.COFFEE:an objective function for multiple sequence alignments[J].Bioinformatics,1998,14(5):407-422.
[3]Katoh K,Toh H.Parallelization of the MAFFT multiple sequence alignment program[J].Bioinformatics,2010,26(15):1899-1900.
[4]Hirosawa M,Hoshida M,Ishikawa M,et al.MASCOT: multiple alignment system for protein sequences based on three-way dynamic programming[J].Bioinformatics,1993,9(2):161-167.
[5]鄒權,郭茂祖,王曉凱,等.基于關鍵字樹的DNA多序列星比對算法[J].電子學報,2009,37(8):1746-1750.
[6]袁激光,金人超,李紅濤.基于A*算法的啟發式算法求解多序列比對問題[J].華中科技大學學報(自然科學版),2003,31(9):50-52.
[7]Kim J,Pramanik S,Chung MJ.Multiple sequence alignment using simulated annealing[J].Bioinformatics,1994,10(4):419-426.
[8]Notredame C,O'Brien EA,Higgins DG.RAGA:RNA sequence alignment by genetic algorithm[J].Nucleic Acids Research,1997,25(22):4570-4580.
[9]張鵬帥,霍紅衛.多序列比對問題的粒子群優化算法求解[J].計算機工程與應用,2005,18:84-87.
[10]Krogh A,Brown M,Mian IS,et al.Hidden Markov models in computational biology:Applications to protein modeling[J].Journal of Molecular Biology,1994,235(5): 1501-1531.
[11]Zou Q,Li XB,Jiang WR,et al.Survey of Map Reduce frame operation in bioinformatics[J].Brief Bioinfor,2014,15(4):637-647.
[12]Zou Q,Shan X,Jiang Y.A novel center star multiple sequence alignment algorithm based on affine gap penalty and K-band[J].Physics Procedia,2012,33:322-327.
[13]Zou Q,Hu Q,Guo M,et al.HAlign:Fast multiple similar DNA/RNA sequence alignment based on the centre star strategy[J].Bioinformatics,2015,31(15):2475-2481.
[14]Needleman SB,Wunsch CD.A general method applicable to the search for similarities in the amino acid sequence of two proteins[J].J Mol Biol,1970,48(3):443-453.
[15]Tanaka M,Cabrera VM,González AM,et al.Mitochondrial genome variation in eastern Asia and the peopling of Japan[J].Genome Res,2004,14(10a):1832-1850.
[16]Zou Q,Mao Y,Hu L,et al.miRClassify:An advanced web server for miRNA family classification and annotation[J].Comput Biol Med,2014,45:157-160.
[17]Wang Q,Wei L,Guan X,et al.Briefing in family characteristics of microRNAs and their applications in cancer research[J].BBA-Proteins Proteom,2014,1844(1):191-197.
[18]Guo L,Chen F.A challenge for miRNA:multiple isomiRs in miRNAomics[J].Gene,2014,544(1):1-7.
[19]Li P,Guo M,Wang C,et al.An overview of SNP interactions in genome-wide association studies[J].Brief Funct Genomics,2015,14(2):143-155.
R730
A
10.11877/j.issn.1672-1535.2016.14.06.03
2015-10-15)
國家自然科學基金(61370010)
(corresponding author),郵箱:zouquan@nclab.net
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學生數理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
