固相萃取-氣質聯用法測定水產品中撲草凈
2016-10-26 10:50:21唐秀范廣宇張云青孟祥龍王海濤徐文科
食品研究與開發 2016年19期
關鍵詞:檢測
唐秀,范廣宇,張云青,孟祥龍,王海濤,徐文科
(連云港出入境檢驗檢疫局,江蘇連云港222042)
固相萃取-氣質聯用法測定水產品中撲草凈
唐秀,范廣宇*,張云青,孟祥龍,王海濤,徐文科
(連云港出入境檢驗檢疫局,江蘇連云港222042)
建立檢測水產品中撲草凈殘留量的固相萃取-氣相色譜質譜聯用法。樣品用乙腈提取,濃縮后經PSA固相萃取柱凈化,由氣質聯用儀選擇離子監測模式下測定。該方法條件下撲草凈在2.5 μg/L~40 μg/L濃度范圍內線性良好,對空白樣品分別添加2、10、20μg/kg撲草凈標準溶液,平均回收率在80%~98%,RSD小于10%,檢出限為0.5μg/kg。
固相萃取;氣相色譜-質譜聯用儀;水產品;撲草凈
撲草凈屬于一種均三氮苯類除草劑,被廣泛應用于大豆、麥類、花生等作物防除雜草,有時也被應用于魚、蝦、貝等養殖水體中的水草及有害藻類的清除[1]。因其化學性質穩定,不易降解,能夠長期存在于環境和生物體中,并且容易隨降水和地表水徑流和滲透作用造成土壤和水體污染,進而進入食物鏈[2]。我國是水產大國,而由于除草劑的廣泛應用造成水產品中撲草凈超標影響了我國水產品的對外出口。2012年,日本查出從我國進口的貝類中多批次撲草凈超標,從而發布多個產品命令檢查通報。日本對撲草凈殘留的應對措施已從紫菜產品擴展到水產品中的貝類[3]。因而建立一種快速準確檢測水產品中撲草凈殘留量的方法來減少出口水產品的檢測周期、提高水產品的通關效率十分必要。
目前,對于環境中撲草凈殘留量檢測的研究較多[4-6],對于水產品中撲草凈的殘留檢測也有少量報道,其中前處理方法有QuEChERS-凝膠滲透色譜法[7]、凝膠滲透色譜-固相萃取法[8]、固相萃取法[9-10]、凝膠滲透色譜法[11]等,采用氣相色譜質譜聯用法[7]、氣相色譜-串聯質譜法[8]、氣相色譜法[9]、液相色譜-串聯質譜法[10-11]等儀器方法測定。凝膠滲透色譜法在高脂肪含量樣品中應用較多,但其儀器昂貴,并且樣品處理耗時較長、使用溶劑量較大。本研究選用固相萃取-氣質聯用法測定水產品中的撲草凈,通過優化前處理方法和儀器的檢測條件,縮短檢測時間,提高檢測效率。
1材料與方法
1.1儀器與試劑
7890A-5975C氣相色譜質譜聯用儀:美國Agilent公司;THZ-92A恒溫振蕩器:上海賀德實驗設備廠;XW-80A旋渦混合器:上海青浦滬西;R215旋轉蒸發儀:瑞士布琪公司;TDL-40C臺式離心機:上海安亭。
色譜純乙腈、丙酮、乙酸乙酯、甲醇:德國merck公司;1 g/6 mL PSA柱、500 mg/6 mL CARBON/PSA、1 g/ 6 mL弗洛里柱、500 mg/6 mL氨基柱、1 g/6 mL中性氧化鋁柱:美國Agilent公司。
撲草凈標準品(純度大于99.0%):德國Dr.公司,準確稱取該標準品,用乙腈定容至100 mL,配制成濃度為100 mg/L的儲備液,使用時用乙腈逐級稀釋。
1.2儀器工作條件
HP-5MS毛細管柱(30 m×0.25 mm×0.25 μm);進樣口溫度250℃;進樣量1 μL;柱流量1 mL/min;不分流進樣;輔助通道溫度280℃;升溫程序為:初始溫度50℃保持2 min,20℃/min升至180℃,10℃/min升至260℃;離子源溫度230℃;四極桿溫度150℃;掃描離子為m/z 241、184、226、199。
1.3樣品前處理
水生動物樣品取可食部分打碎,稱取5.0 g于錐形瓶中,加入乙腈50 mL混勻,放入振蕩器中振蕩提取0.5 h。加入適量氯化鈉混勻靜置,提取液經無水硫酸鈉過濾于平底燒瓶中,用乙腈分3次清洗錐形瓶內殘渣過濾于同一燒瓶中,于40℃旋轉蒸發至干。
用3 mL丙酮+正己烷(1∶4,體積比)混合溶液溶解以上殘渣,將PSA柱用6 mL正己烷清洗活化,然后將以上溶液加入PSA柱內,再用丙酮+正己烷(1∶4,體積比)混合溶液2 mL分二次清洗燒瓶并過柱,收集的洗脫液于40℃水浴中氮吹至干。用1 mL乙腈定容,3 500 r/min下離心5 min,取上清液過0.22 μm濾膜后上機檢測。
2結果與討論
2.1儀器方法選擇
根據標準和參考文獻,考慮撲草凈與雜質有效分離,并保證高沸點雜質能流出色譜柱,選取了氣相色譜升溫程序,在1.2所述條件下,撲草凈保留時間為12.99 min,撲草凈總離子流圖見圖1。
根據撲草凈全掃描質譜圖確定撲草凈的定量離子為m/z 241,定性離子為m/z 184、226、199。
2.2提取溶劑選擇

圖1 撲草凈總離子流圖Fig.1Total ion chromatogram of prometryn
農藥殘留測定前處理常用的提取劑主要有乙腈、丙酮、乙酸乙酯和甲醇,試驗分別對4種提取劑的提取效果進行對比,結果發現丙酮和甲醇會提取出較多的水分,導致提取液旋轉蒸發濃縮時無法完全蒸干,從而影響提取效率,而用乙酸乙酯提取不如用乙腈提取的效率高,經過對比,試驗選用乙腈作為提取溶劑。
2.3固相萃取柱選擇
常用的固相萃取柱有PSA柱、弗洛里柱、氨基柱和中性氧化鋁柱,試驗對比了4種固相萃取柱的凈化效果,結果發現回收效率均能達到80%以上,但弗洛里柱和中性氧化鋁的基線較高,容易受到干擾,而氨基柱凈化效果不理想,干擾峰與撲草凈沒有達到基線分離,故最終選擇了PSA柱。
2.4洗脫溶液選擇
試驗以丙酮和正己烷的混合溶液為洗脫液,分別配制丙酮和正己烷的體積比為1∶9、2∶8、3∶7、4∶6和5∶5的混合溶液,數據見表1。
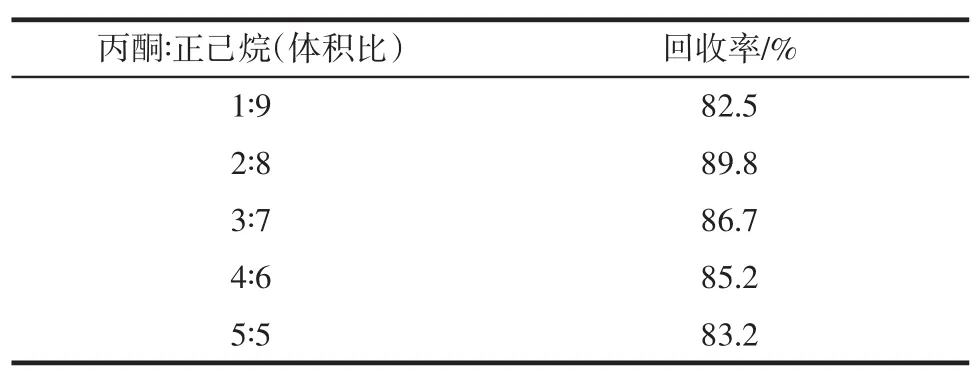
表1 洗脫溶劑影響Table 1Effect of elution solvent
表1結果顯示,丙酮和正己烷的體積比為2∶8時回收率達到最大,隨著丙酮體積比例的增加色譜峰的基線不斷升高,添加回收率也隨之下降。最終選用丙酮與正己烷體積比為2∶8的混合溶液為洗脫液。
2.5洗脫體積的選擇
洗脫溶液的用量為剛好能把目標物質洗脫下來為宜,過多使用反而會引入其他干擾,在確定洗脫混合溶液比例的基礎上,分別對5個不同體積洗脫液的洗脫效率進行了對比,數據見表2。

表2 洗脫體積影響Table 2Effect of elution volume
試驗結果顯示當洗脫液體積達到5 mL時即可將大部分目標物質洗脫出來,隨后隨著洗脫體積的增加目標物質的回收率并沒有增加而且色譜峰的干擾變大,由此確定洗脫體積為5 mL。
2.6方法的分析特性
配制標準系列,在選定的儀器條件下進行測定,以濃度為橫坐標,響應值峰面積為縱坐標繪制標準曲線,曲線方程為A=176.2C-83.04,線性相關系數為0.999 8,線性范圍為2.5 μg/L~40 μg/L,根據3倍信噪比計算出本試驗方法的檢出限為0.5 μg/kg。
稱取空白貽貝肉樣品各5 g,分別添加2、10、20 μg/kg標準溶液,每個加標水平平行測定6次,按照選定的實驗條件和儀器條件得到的添加回收率和測定值的相對標準偏差(RSD)見表3。

表3 添加回收率和精密度(n=6)Table 3Recoveries and precision of the method(n=6)
2.7實際樣品分析
試驗對文蛤、河蜆、赤貝、蝦、貽貝肉等11種樣品進行了測定,其中10個樣品檢測值低于2 μg/kg,1個皺紋蛤樣品中的撲草凈檢出量33 μg/kg,超出標準限量10 μg/kg。
3結論
本研究建立了一種固相萃取-氣相色譜質譜聯用法測定水產品中撲草凈殘留量的方法。樣品采用乙腈提取,PSA固相萃取柱凈化,由氣質聯用儀檢測和確證,外標法定量。該方法條件下撲草凈在2.5 μg/L~40 μg/L濃度范圍內線性關系良好,通過向空白水產品樣品中添加不同濃度水平的撲草凈標準溶液,平均回收率在80%~98%,相對標準偏差(n=6)為2.7%~8.6%,方法檢出限為0.5 μg/kg。該方法具有操作簡單、定性準確、靈敏度高的特點,可以應用于進出口水產品中撲草凈的檢測。
水產品中撲草凈的殘留問題已經影響到我國對日本和歐盟出口,應該引起水產養殖和出口加工企業關注。撲草凈檢出率高有可能是養殖水體污染或者在除去養殖中的雜草過量使用撲草凈,相關企業可以分析具體原因妥善應對。
[1]張騫月,吳偉.撲草凈在養殖水體中的生態毒理效應及其微生物降解的研究進展[J].生物災害科學,2014,37(1):64-69
[2]董麗嫻,陳玲,李建華,等.水中三嗪類除草劑的檢測與分析質量控制[J].安全與環境學報,2006,6(5):35-38
[3]李慶鵬,秦達,崔文慧,等.我國水產品中農藥撲草凈殘留超標的警示分析[J].食品安全質量檢測學報,2014,5(1):108-112
[4]李衛建,聶志強,蔡彥明,等.氣相色譜法同時測定土壤中13種三嗪類除草劑殘留量的方法研究[J].農業環境科學學報,2009,28(1):211-215
[5]曹軍,尹小樂,布文安,等.環境中除草劑撲草凈殘留分析方法的研究[J].分析科學學報,2007,23(4):397-400
[6]余晟,黃克靖,余萌,等.固相萃取-高效液相色譜法同時檢測水樣中戊唑醇、乙霉威、晴菌唑、精甲霜靈和撲草凈5種農藥殘留[J].分析化學,2012,40(7):1065-1070
[7]孫曉杰,郭萌萌,孫偉紅,等.QuEChERS在線凝膠色譜-氣相色譜/質譜快速檢測水產品中農藥多殘留[J].分析科學學報,2014,30(6):868-872
[8]張華威,劉慧慧,田秀慧,等.凝膠色譜-固相萃取-氣相色譜-串聯質譜法測定水產品中9種三嗪類除草劑[J].質譜學報,2015,36(2):177-184
[9]宋業萍,宗萬里,于忠飛,等.氣相色譜法測定貝類中撲草凈的殘留量[J].衛生研究,2014,43(5):790-792
[10]宗萬里,劉新才,宋業萍,等.液相色譜-串聯質譜法測定花色蛤中撲草凈殘留量[J].農產品質量與安全,2014(4):52-55
[11]劉棟,李蓉娟,陳曉東,等.GPC-HPLC-MS/MS法檢測貝類中撲草凈殘留[J].食品研究與開發,2013,34(24):205-208
Determination of Prometryn in Aquatic Products by Solid Phase Extraction-Gas Chromatography Mass Sepectrometry
TANG Xiu,FAN Guang-yu*,ZHANG Yun-qing,MENG Xiang-long,WANG Hai-tao,XU Wen-ke
(Lianyungang Entry-exit Inspection and Quarantine Bureau,Lianyungang 222042,Jiangsu,China)
A method for the determination of prometryn in aquatic products with solid phase extraction-gas chromatography mass sepectrometry(SPE/GC-MS)was established.The prometryn in samples was extracted with acetonitrile,and the supernatant was purified by primary secondary amine(PSA)solid phase column. Then,the analyte was determined in selected ion monitoring of GC-MS.The calibration curves of prometryn showed good linearity in 2.5 μg/L-40 μg/L.The mean recoveries of the prometryn varied from 80%to 98% when spiked at 2,10,20 μg/kg,and the relative standard deviations were less than 10%.The limit of the quantitative was 0.5 μg/kg.
solidphase extraction(SPE);gas chromatography-mass spectrometry(GC-MS);aquatic products;prometryn
10.3969/j.issn.1005-6521.2016.19.033
2015-11-12
連云港市科技公共服務平臺資助項目(JC1404)
唐秀(1986—),女(漢),助理工程師,學士,主要從事進出口食品中農藥殘留檢測工作。
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
