快速溶劑萃取-HPLC法測定骨碎補中柚皮苷的含量
2016-11-24 09:48:49黃北雄戴柏桉廣西梧州食品藥品檢驗所廣西梧州543002
中國藥房 2016年27期
黃北雄,戴柏桉(廣西梧州食品藥品檢驗所,廣西梧州 543002)
快速溶劑萃取-HPLC法測定骨碎補中柚皮苷的含量
黃北雄*,戴柏桉(廣西梧州食品藥品檢驗所,廣西梧州543002)
目的:為骨碎補中柚皮苷的提取及測定開辟新的途徑。方法:采用ASE350型快速溶劑萃取系統提取骨碎補中的柚皮苷;以高效液相色譜法測定骨碎補中柚皮苷的含量。色譜柱為Kinetex XB-C18,流動相為甲醇-5%乙酸(25∶75,V/V),流速為0.8 ml/min,檢測波長為283 nm,柱溫為40℃,進樣量為10μl。結果:柚皮苷的進樣量線性范圍為0.073 0~0.730 0μg(r=0.999 9));定量限為0.73 μg,檢測限為0.022 μg;精密度、穩定性、重復性試驗的RSD<2%;加樣回收率為98.92%~100.85%(RSD=0.72%,n=6)。結論:該方法簡便、準確、專屬性強,可用于測定骨碎補中柚皮苷的含量。
高效液相色譜法;骨碎補;柚皮苷;快速溶劑萃取
骨碎補為水龍骨科植物槲蕨Drynaria fortune(Kunze)J. Sm.的干燥根莖,氣微、味淡、微澀,有療傷止痛、補腎強骨、外用消風祛斑的功效,可用于跌撲閃挫、筋骨折傷、腎虛腰痛、筋骨痿軟、耳鳴耳聾、牙齒松動、外治斑禿、白癜風等的治療[1]。現代藥理研究也表明骨碎補具有降血脂、降低胺基糖苷類抗生素的毒性和促進骨吸收等作用[2]。柚皮苷是骨碎補的主要有效成分之一,2015年版《中國藥典》(一部)以柚皮苷作為其質量評價的指標。目前,骨碎補中柚皮苷的提取主要有煎煮法、加熱回流法、索氏提取法[3-4]、超聲萃取法、微波輔助萃取法[5-8],測定方法有高效液相色譜法(HPLC)[9-11]和薄層-紫外分光光度法[12-13],但是上述方法提取時間長、測定步驟煩瑣。因此,筆者利用美國Dionex公司的快速溶劑萃取系統對樣品進行提取,縮短提取時間、提高效率,并采用HPLC法測定骨碎補中柚皮苷的含量,以期為骨碎補中柚皮苷的提取及測定開辟新的途徑。
1 材料
1.1儀器
ASE350型快速溶劑萃取儀(美國Dionex公司);Ultimate 3000 DGLC型HPLC儀(美國Thermo Fisher Scientific公司);XA205DU型十萬分之一電子天平(瑞士Mettler-Toledo公司)。
1.2試劑
柚皮苷對照品(中國食品藥品檢定研究院,批號:200001,純度:94.7%);甲醇為色譜純,其余試劑均為分析純,水為超純水。
1.3藥材
骨碎補(玉林市康華中藥飲片有限公司,批號:131206;廣西玉林市祥生中藥飲片有限責任公司,批號:140601;玉林市華濟中藥飲片有限公司;批號:14070301)經廣西梧州食品藥品檢驗所葉小強藥師鑒定為水龍骨科植物槲蕨Drynaria fortune(Kunze)J.Sm.的干燥根莖。
2 方法與結果
2.1色譜條件
色譜柱:Kinetex XB-C18(100 mm×4.6 mm,2.6 mm);流動相:甲醇-5%乙酸(25∶75,V/V);流速:0.8 ml/min;檢測波長:283 nm;柱溫:40℃;進樣量:10μl。
2.2溶液的制備
2.2.1對照品溶液精密稱取柚皮苷對照品適量,置于25 ml量瓶中,加甲醇制成每1 ml含柚皮苷36.5 μg的對照品溶液。
2.2.2供試品溶液取樣品粗粉0.1 g,按1∶1的比例加入硅藻土后混勻,裝于10 ml萃取池中,再用硅藻土填滿萃取池,在ASE350型快速溶劑萃取儀控制面板編程進行萃取試驗(萃取壓力為1 700 psi,溶劑為甲醇,萃取溫度為120℃,循環次數為2次,萃取時間為5 min,沖洗體積為100%)。萃取結束后,將萃取液轉移至50 ml量瓶中,加甲醇定容,搖勻,濾過,取續濾液,即得。
2.3系統適用性試驗
精密量取“2.2”項下對照品溶液、供試品溶液各適量,按“2.1”項下色譜條件進樣測定,記錄色譜,詳見圖1。由圖1可知,在該色譜條件下,各成分均能達到基線分離,分離度>1.5;理論板數以柚皮苷峰計為5 000;保留時間為5.73 min。結果表明,其他成分對測定無干擾。
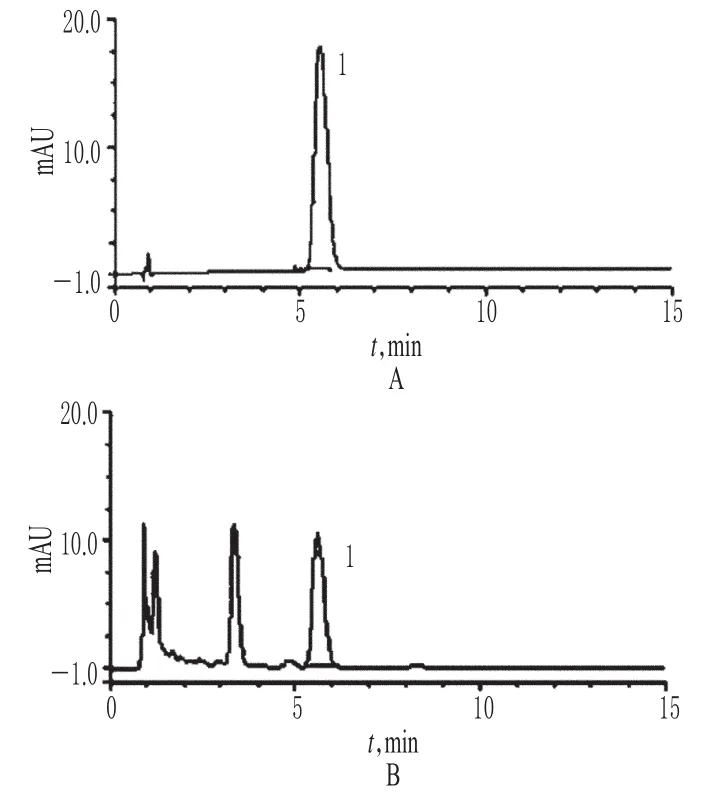
圖1 高效液相色譜圖A.對照品;B.供試品;1.柚皮苷Fig 1 HPLC chromatogramsA.reference substance;B.test sample;1.naringin
2.4快速萃取條件的確定
2.4.1靜態萃取溫度平行取樣品粗粉適量,共5份,按1∶1的比例加入硅藻土后混勻,裝于10 ml萃取池中,用硅藻土填滿萃取池,參考2015年版《中國藥典》(一部)“骨碎補含量測定項下”水浴的溫度,分別設定提取溫度為80、100、120、140、160℃進行提取,再按“2.1”項下色譜條件進樣測定并計算柚皮苷的含量。結果,120℃時柚皮苷的提取量最高,超過120℃以后柚皮苷的提取量呈遞減趨勢,同時色譜圖中未知雜質成分增多。因此,本試驗選擇120℃為提取溫度。
2.4.2循環次數平行取樣品粗粉適量,共3份,按1∶1的比例加入硅藻土后混勻,裝于10 ml萃取池中,用硅藻土填滿萃取池,分別設定循環次數為1、2、3次進行提取,再按“2.1”項下色譜條件進樣測定并計算柚皮苷的含量。結果,循環次數為2次和3次時柚皮苷的提取量最高。為節約提取時間,故本試驗的循環次數選擇2次。
2.4.3靜態萃取時間平行取樣品粗粉適量,共4份,按1∶1的比例加入硅藻土后混勻,裝于10 ml萃取池中,用硅藻土填滿萃取池,分別設定靜態萃取時間為3、5、7、9 min進行提取,再按“2.1”項下色譜條件進樣測定并計算柚皮苷的含量。結果,提取時間為5、7、9 min時,柚皮苷的含量最高。為節約試驗時間,故本試驗選擇靜態萃取時間為5 min。
2.4.4沖洗體積平行取樣品粗粉適量,共6份,按1∶1的比例加入硅藻土后混勻,裝于10 ml萃取池中,用硅藻土填滿萃取池,分別設定沖洗體積為0、40%、80%、100%、120%、150%進行提取,再按“2.1”項下色譜條件進樣測定并計算柚皮苷的含量。結果,沖洗體積為100%、120%、150%時柚皮苷的含量最高。為節約試驗成本,故本試驗沖洗體積選擇為100%。
2.5線性關系考察
分別精密吸取“2.2.1”項下對照品溶液0、2、5、10、15、20 μl,按“2.1”項下色譜條件進樣測定,記錄峰面積。以進樣量(x,μg)為橫坐標、峰面積(y)為縱坐標進行線性回歸,得柚皮苷的回歸方程為y=1 888.8x+8.980 5(r=0.999 9)。結果表明,柚皮苷的進樣量線性范圍為0.073 0~0.730 0μg。
2.6定量限與檢測限考察
取“2.2.1”項下對照品溶液適量,等倍逐步稀釋,按“2.1”項下色譜條件連續進樣測定6次,記錄峰面積。當信噪比為10∶1時,得柚皮苷的定量限為0.73 μg;當信噪比為3∶1時,得柚皮苷的檢測限為0.022 μg。
2.7精密度試驗
取“2.2.1”項下對照品溶液適量,按“2.1”項下色譜條件進樣測定,記錄峰面積。結果,柚皮苷峰面積的RSD=0.04%(n=6),表明儀器精密度良好。
2.8穩定性試驗
取“2.2.2”項下供試品溶液適量,分別于室溫下放置0、2、4、6、8、10、12 h時按“2.1”項下色譜條件進樣測定,記錄峰面積。結果,柚皮苷峰面積的RSD=0.99%(n=7),表明供試品溶液在室溫下12 h內穩定性良好。
2.9重復性試驗
取樣品(批號:131206)粗粉適量,按“2.2.2”項下方法平行制備6份供試品溶液,再按“2.1”項下色譜條件進樣測定,記錄峰面積。結果,柚皮苷峰面積的RSD=1.47%(n=6),表明本方法重復性良好。
2.10加樣回收率試驗
取已知含量的樣品(批號:131206)粗粉適量,共6份,每份0.05 g,精密稱定,分別精密加入一定質量的柚皮苷對照品,按“2.2.2”項下方法制備6份供試品溶液,再按“2.1”項下色譜條件進樣測定,記錄峰面積并計算加樣回收率,結果詳見表1。
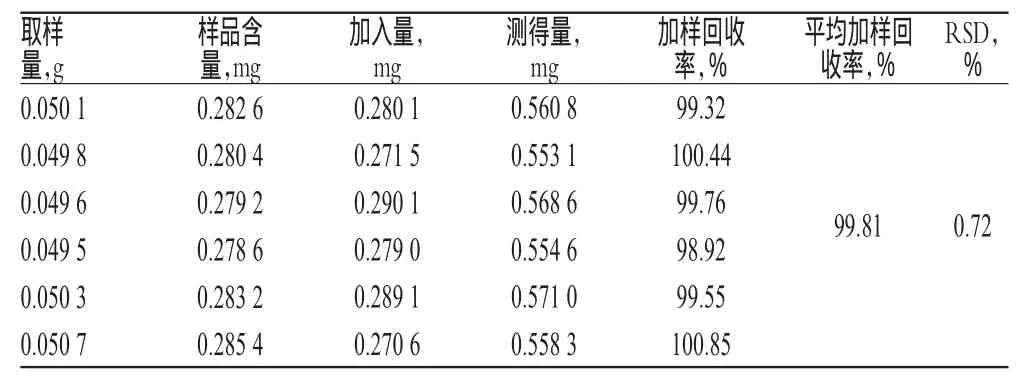
表1 加樣回收率試驗結果(n=6)Tab 1 Results of recovery test(n=6)
2.11樣品含量測定
取3批樣品粗粉適量,按“2.2.2”項下方法制備供試品溶液,再按“2.1”項下色譜條件進樣測定,記錄峰面積并計算柚皮苷的含量,結果詳見表2。
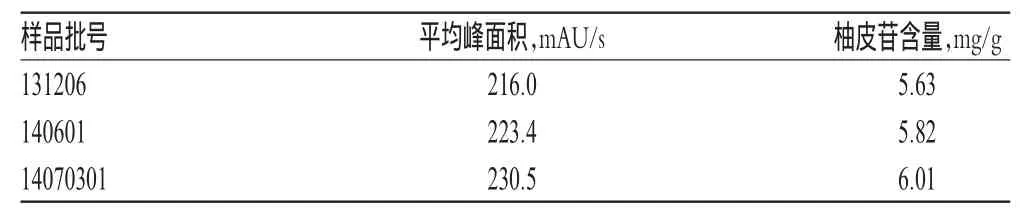
表2 樣品含量測定結果(n=3)Tab 2 Results of content determination of samples(n=3)
3 討論
本試驗比較了甲醇-乙酸-水(35∶4∶65,V/V/V)、甲醇-5%乙酸(25∶75,V/V)兩種不同比例的流動相,通過預試驗表明,甲醇-5%乙酸(25∶75,V/V)為流動相時,柚皮苷峰與相鄰色譜峰的分離效果較好、峰型較好。故本試驗選擇甲醇-5%乙酸(25∶75,V/V)為流動相。
綜上所述,本方法簡便、準確、專屬性強,可用于測定骨碎補中柚皮苷的含量。
[1]國家藥典委員會.中華人民共和國藥典:一部[S].2015年版.北京:中國醫藥科技出版社,2015:256.
[2]劉振麗,呂愛平,張秋海,等.骨碎補脂溶性成分研究[J].中國中藥雜志,1999,24(4):222.
[3]張琳,楊中林.水提法與醇提法對骨碎補中總黃酮含量的影響比較[J].江蘇藥學與臨床研究,2004,12(3):30.
[4]伍奕,蔣曉煌,蔣孟良,等.骨碎補中柚皮苷含量測定方法的優化研究[J].湖南中醫藥大學學報,2010,30(3):48.
[5]方婧,楊洪軍,付梅紅,等.微波協助提取在中藥飲片含量測定中的應用(4):微波法與藥典法測定骨碎補中柚皮苷含量比較[J].中國實驗方劑學雜志,2012,18(6):75.
[6]高軍,劉富春.正交試驗優選枳實中柚皮苷的提取工藝[J].中國藥房,2014,25(35):3 291.
[7]李園園,石磊,張振巍,等.混合均勻設計結合響應面法優選枳苓六妙口服液中藥材的提取工藝[J].中國藥房,2013,24(39):3 685.
[8]高穎,房德敏,王巨存,等.蘇氏接骨膠囊中骨碎補和菟絲子的質量控制[J].中國醫院藥學雜志,2010,30(4):333.
[9]張紅旭,郭輝.HPLC法測定骨碎補酊中柚皮苷含量[J].西北藥學雜志,2006,21(2)63.
[10]岳春華,李順祥.從骨碎補中制備新北美圣草苷和柚皮苷對照品的研究[J].中草藥,2008,39(4):529.
[11]李遇伯,孟繁浩,潘曉峰,等.HPLC同時測定骨碎補藥材中新北美圣草苷和柚皮苷的含量[J].藥物分析雜志,2006(6):808.
[12]呂勇均,邱宗蔭.中藥骨碎補提取物質量標準研究[J].中國藥業,2007,16(23):26.
[13]陳潔.薄層-紫外分光光度法測定骨碎補中柚皮甙的含量[J].中醫正骨雜志,2007,19(3):76.
(編輯:劉柳)
Determination of Naringin in Davallia mariesii byAccelerated Solvent Extration-HPLC
HUANG Beixiong,DAI Baian(Guangxi Wuzhou Institute for Food and Drug Control,Guangxi Wuzhou 543002,China)
OBJECTIVE:To develop a new way for the extration and determination of naringin in Davallia mariesii.METHODS:Naringin in D.mariesii was extracted by ASE350 accelerated solvent extraction system;HPLC was performed for the content determination of naringin in D.mariesii,the column was Kinetex XB-C18with mobile phase of methanol-5%acetic acid(25∶75,V/V)at a flow rate of 0.8 ml/min,detection wavelength was 283 nm,column temperature was 40℃,and injection volume was 10 μl.RESULTS:The linear range of naringin was 0.073 0-0.730 0μg(r=0.999 9);the limit of quantitation was 0.73 μg,the limit of detection was 0.022 μg;RSDs of precision,stability and reproducibility tests were lower than 2%;recovery was 98.92%-100.85%(RSD=0.72%,n=6).CONCLUSIONS:The method is simple,accurate and specific,and can be used for the content determination of naringin in D.Mariesii.
HPLC;Davallia mariesii;Naringin;Accelerated solvent extraction
R917
A
1001-0408(2016)27-3875-03
10.6039/j.issn.1001-0408.2016.27.44
*副主任藥師。研究方向:食品藥品檢驗。電話:0774-3886969。E-mail:392271494@qq.com
(2015-12-19
2016-07-16)
