碳納米管上原位沉積鈀-鈷-鎳納米顆粒及其對乙醇氧化的電活性
2016-12-05 05:42:11陳清華易清風周秀林劉小平聶會東徐國榮
無機化學學報 2016年7期
關鍵詞:催化劑
陳清華 易清風*,,2 陽 錚 周秀林 劉小平 聶會東 徐國榮
(1湖南科技大學化學化工學院,湘潭411201)
(2理論有機化學與功能分子教育部重點實驗室,湘潭411201)
碳納米管上原位沉積鈀-鈷-鎳納米顆粒及其對乙醇氧化的電活性
陳清華1易清風*,1,2陽錚1周秀林1劉小平1聶會東1徐國榮1
(1湖南科技大學化學化工學院,湘潭411201)
(2理論有機化學與功能分子教育部重點實驗室,湘潭411201)
以水合肼為還原劑,在水和乙醇的混合溶液中制備多壁碳納米管(MWCNT)負載的納米鎳(Ni/MWCNT)和納米鎳鈷(Ni-Co/ MWCNT)顆粒,然后將它們分別與氯化鈀溶液反應,形成的鈀納米顆粒原位沉積在MWCNT表面,從而得到MWCNT負載的Pd-Ni/MWCNT和Pd-Ni-Co/MWCNT催化劑。SEM和TEM圖像顯示,MWCNT上的催化劑顆粒是由5~10 nm的小顆粒團聚而成的30~100 nm的大顆粒,三金屬催化劑的粒徑比雙金屬的粒徑小,在MWCNT上的分散度更高。ICP和EDS分析顯示,Pd直接還原并包覆在納米鎳和納米鎳鈷表面;采用循環伏安和計時電流技術,研究了催化劑在堿性溶液中對乙醇氧化的電催化活性,結果表明,Pd-Ni-Co/MWCNT催化劑對乙醇氧化具有強的電催化活性,乙醇氧化對應的峰電流密度達101.8 mA·cm-2,并且催化劑催化活性穩定。
鈀催化劑;乙醇氧化;鎳納米顆粒;燃料電池;原位還原
0 引言
直接醇燃料電池具有能量密度高、燃料易存儲且安全、低毒等優點,受到各國科學家廣泛研究。直接乙醇燃料電池以乙醇為燃料,它是一種生物質燃料,能量密度高,污染排放物少,是一種環境友好和可再生型的燃料,因此研究對乙醇電化學氧化具有高效活性的催化劑具有重要的實際意義[1]。雖然鉑或鉑基催化劑對乙醇氧化具有優異的活性,但鉑的儲量和成本以及易毒化限制了它的廣泛使用[2-4]。研究表明,鈀在堿性環境下能夠高效催化乙醇氧化,并且鈀在地球上的豐度至少是鉑的50倍,因此,制備對乙醇氧化更加高效的鈀基催化劑具有重要的研究意義[5-6]。在鈀催化劑中摻入過渡金屬或其氧化物可以提高催化劑的活性及穩定性,并能減少燃料電池的成本[7-10],如PdPt/CNTs[11],PdCo[12]和PdCu/C[13]等;此外,Ni和Co可以提高Pd在載體表面的分散性,減少金屬的團聚,提高Pd的催化活性和其抗毒性[14-17]。通過“雙金屬原理”和“配位效應”可以較合理地解釋這類復合鈀基催化劑所具有的強催化活性[18-20]。
本文首先以氯化鎳(NiCl2·6H2O)和氯化鈷(CoCl2·6H2O)為金屬前驅體,以水合肼為還原劑將金屬離子還原[21],得到的納米顆粒負載在碳納米管表面,制備納米鎳和納米鎳鈷顆粒修飾的碳納米管復合物(Ni/MWCNT和NiCo/MWCNT);然后以此復合物為還原劑將Pd2+還原為金屬Pd顆粒[22],形成的Pd顆粒原位負載在復合物表面,從而形成PdNi/ MWCNT和PdNiCo/MWCNT復合催化劑。利用SEM、TEM和XRD等技術對催化劑的表觀結構進行了分析;采用循環伏安和計時電流法,對催化劑在堿性溶液中電化學催化乙醇氧化的活性和穩定性進行了研究。結果表明,催化劑的鈀負載量小,但具有優異的電化學活性。
1 實驗部分
1.1實驗試劑與儀器
本文所用試劑為氯化鈀(PdCl2,天津市科密歐化學試劑有限公司),氯化鎳(NiCl2·6H2O,廣州市海珠區華安化工廠),氯化鈷(CoCl2·6H2O,汕頭市光華化學廠有限公司),無水乙醇(CH3CH2OH,天津市大茂化學試劑廠),氫氧化鈉(NaOH,天津市永大化學試劑有限公司),水合肼(N2H4·H2O,80%,湖南匯虹試劑有限公司)等,所有試劑均為分析純,未對其進行后續提純;多壁碳納米管(MWCNT)管徑為20~30 nm;所用純水為二次蒸餾水。
實驗儀器:XRD衍射儀(UltimaⅣMultipurpose X-Ray Diffraction System,Rigaku,Japan),輻射源為Cu Kα(λ=0.154 18 nm),管電壓為40 kV,管電流為40mA,掃描速率為5°·min-1,掃描范圍為20°~90°;場發射掃描電鏡(Nova NanoSEM230),加速電壓為15 kV;透射電子顯微鏡(Tecnai G20,FEICompany,USA),加速電壓為200 kV;等離子體發射光譜儀(ICPS-7510,日本島津株式會社)。Autolab PGSTAT30(荷蘭)電化學工作站。
1.2催化劑的制備
1.2.1Ni/MWCNT納米顆粒的制備
首先將MWCNT在濃H2SO4和濃HNO3的混合液中,于80℃水浴回流反應4 h,離心分離后過濾,并用大量水洗至弱酸性,真空40℃干燥,得到酸化MWCNT。然后將165.9mg NiCl2·6H2O、200.0 mg酸化MWCNT與13.6 mL無水乙醇和16.7 mL水混合,攪拌均勻后置于50℃水浴鍋中,不斷攪拌;同時用5.0 mol·L-1NaOH調節pH=14左右,隨后逐滴加入新配置的20 mL 10%N2H4·H2O,50℃下水浴保溫0.5 h。所得樣品用無水乙醇和二次水洗至中性,真空40℃干燥4 h。
1.2.2NiCo/MWCNT納米顆粒的制備
按物質的量之比nNi∶nCo=1∶1加入NiCl2·6H2O和CoCl2·6H2O,在與Ni/MWCNT相同的制備條件下得到NiCo/MWCNT納米顆粒。
1.2.3PdNi/MWCNT和PdNiCo/MWCNT催化劑的制備
分別將Ni/MWCNT和NiCo/MWCNT顆粒于水中超聲分散30 min,另將5 mmol·L-1PdCl2用稀NaOH溶液調節pH=3~4。按物質的量之比nPd∶nNi=1∶2和nPd∶(nNi+nCo)=1∶2的比例,分別將已調好pH的PdCl2溶液緩慢滴加到上述Ni/MWCNT和NiCo/ MWCNT在水中的分散液中。攪拌過夜后抽濾,并用水洗至中性,40℃下真空干燥10 h,得到的催化劑分別記為PdNi/MWCNT和PdNiCo/MWCNT。作為對比,在相同條件下以NaBH4為還原劑制備了Pd負載量為20%的Pd/MWCNT催化劑。
1.3樣品測試
電化學性能測試在常規的三電極體系中進行,參比電極為Hg/HgO(1.0 mol·L-1NaOH),輔助電極為鉑電極,工作電極為催化劑修飾的玻碳電極(d=3 mm)。測試前對玻碳電極(GC)進行打磨拋光,超聲清洗3min后自然晾干備用。之后在950μL無水乙醇和50μL Nafion(5%)的混合溶液中加入5 mg催化劑,超聲分散1 h至形成均勻的懸濁液,移取12 μL懸濁液滴在GC電極表面,室溫干燥約3 h,得到催化劑修飾的工作電極。本文所報道的電位均相對于Hg/HgO,電流密度均相對于玻碳電極的幾何面積,電化學測試所用電解質均為1.0mol·L-1NaOH或含乙醇的1.0 mol·L-1NaOH,循環伏安(CV)測試電位控制在0.9到0.5 V,掃描速度為50mV·s-1。恒電位階躍電位為-0.4 V。
2 結果與討論
圖1分別為Ni/MWCNT(a,a′)和NiCo/MWCNT (b,b′)的SEM和TEM圖,從SEM圖(圖1a,b)中看出,MWCNT纏繞到所形成的金屬納米顆粒的結構中,Ni和NiCo顆粒比較大,有團聚現象,但NiCo的粒徑比Ni小,Ni和NiCo顆粒分別約為250和150 nm,原因可能是Ni為磁性金屬,容易相互吸引,比較容易導致顆粒之間的團聚;而有Co存在時,減少了Ni納米粒子的相互接觸而使團聚現象減少。但從催化劑的TEM圖(圖1a′,b′)可看到,在MWCNT的管壁上負載了大量的金屬顆粒,粒徑大小分別為8~15和8~13 nm,也說明了SEM圖像中的團聚現象是由較小的顆粒形成的。
Pd/MWCNT,PdNi/MWCNT和PdNiCo/MWCNT的SEM和TEM圖見圖2,從SEM圖看到,3種催化劑顆粒均較均勻地負載在MWCNT上,Pd/MWCNT、PdNi/MWCNT和PdNiCo/MWCNT的催化劑顆粒分別為4~6、60~100和30~50 nm,說明Pd顆粒的粒徑比較小,分散性更好,且大部分顆粒大小均勻,僅有少量的團聚。從圖2(b)和(c)看出,雙金屬PdNi/ MWCNT和三金屬PdNiCo/MWCNT復合催化劑顆粒比純鈀的大,但PdNiCo/MWCNT的顆粒比PdNi/ MWCNT的顆粒小,且均比Ni/MWCNT和NiCo/ MWCNT的顆粒小,這是Ni和NiCo與Pd2+進行還原反應的結果。從3種催化劑的TEM圖中可知,催化劑都有少量的團聚現象,顆粒大小分別在4~6、8~10和5~8 nm。從圖2(b′)和圖2(c′)得出的顆粒大小數據與圖2(b)和(c)不同,說明催化劑PdNi/MWCNT和PdNiCo/MWCNT是由許多小顆粒形成的。
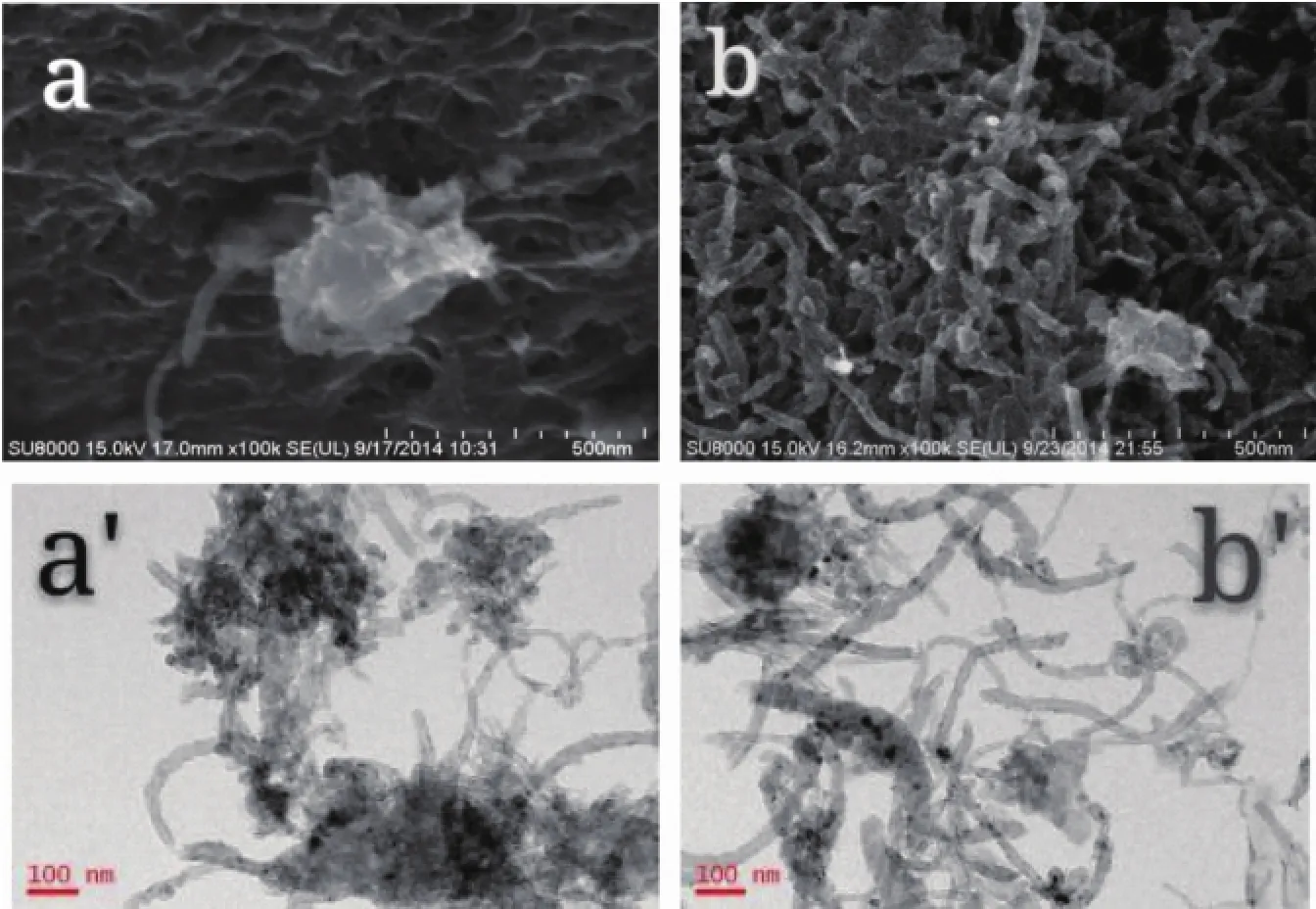
圖1 Ni/MWCNT(a,a′)和NiCo/MWCNT(b,b′)的SEM(a,b)和TEM(a′,b′)圖Fig.1 SEM(a,b)and TEM(a′,b′)images of Ni/MWCNT(a,a′)and NiCo/MWCNT(b,b′)
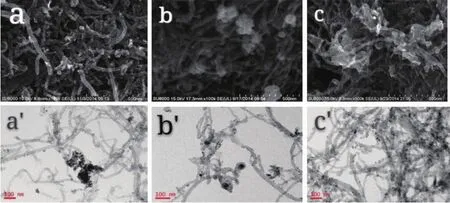
圖2 Pd/MWCNT(a,a′),PdNi/MWCNT(b,b′)和PdNiCo/MWCNT(c,c′)的SEM(a,b,c)和TEM(a′,b′,c′)圖Fig.2 SEM(a,b,c)and TEM(a′,b′,c′)images of Pd/MWCNT(a,a′),PdNi/MWCNT(b,b′)and PdNiCo/MWCNT(c,c′)
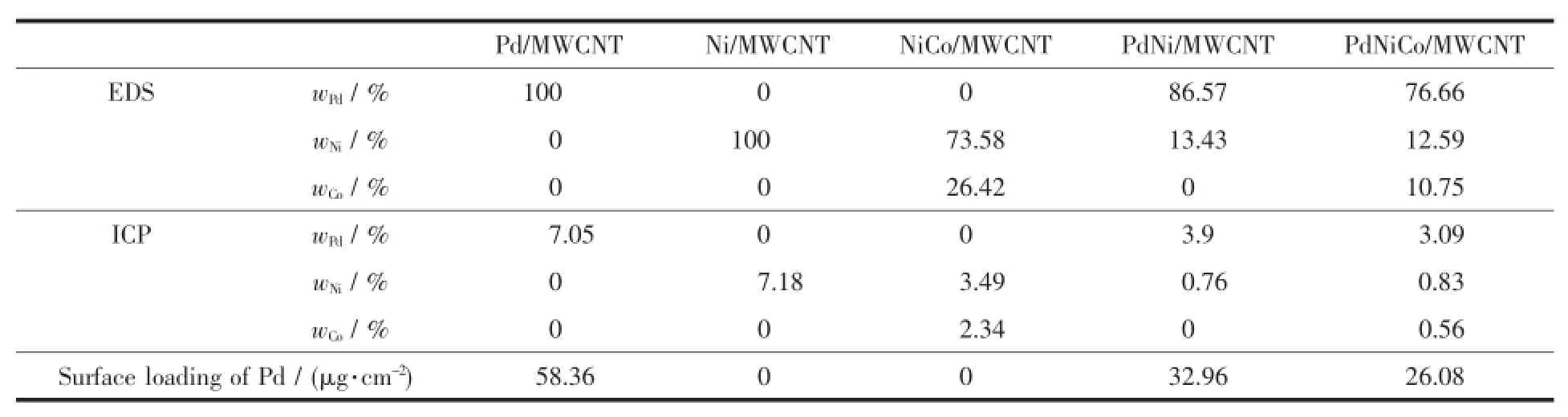
表1 不同催化劑的金屬元素質量分數及電極表面Pd質量分數Table1 M ass percentage of the as-prepared catalysts and Pd loading on electrode of the prepared catalysts
催化劑表面上和催化劑整體中的金屬含量分別用EDS和ICP技術進行了分析,結果見表1。EDS是對催化劑表面微區所激發的體積為10μm3左右所含各種元素含量的半定量分析,ICP是對催化劑中某種元素含量的定量分析。從表1看出,在制備NiCo/MWCNT的反應投料比為1:1,而實際反應后負載在MWCNT上的Co與Ni的質量比并不等于1,這是由于在pH較高時Co2+易形成氫氧化物而不易被還原成金屬單質,可能在反應開始就形成的氫氧化物或氧化物被后來還原的Ni覆蓋,或是在后續處理過程中流逝。從EDS分析可知,PdNi/ MWCNT和PdNiCo/MWCNT的Pd/Ni質量比分別為6.45和6.09,而ICP分析得到的Pd/Ni質量比分別為5.13和3.72,以此可知大部分Pd顆粒沉積在Ni顆粒和NiCo顆粒表面,是以原位沉積的方式進行反應的,這樣增大了金屬之間的接觸面,Pd的活性位點有可能增多。表1顯示,純鈀電極表面的含鈀量最大,說明用NaBH4能很好地將Pd2+還原為Pd。Ni和Co的還原能力沒有NaBH4強,使得反應并未按最初的投料比進行,與EDS分析結論相似。
圖3為催化劑的XRD圖,所有催化劑在衍射角為25.8°的衍射峰對應于MWCNT的(002)晶面。除Pd/MWCNT之外,其它催化劑在44.5°、51.9°和76.3°的衍射峰分別對應于Ni晶體的(111)、(200)和(220)面心立方晶體結構。Pd/MWCNT、PdNi/MWCNT和PdNiCo/MWCNT在40.1°、44.6°、68.1°和82.1°的衍射峰分別對應于Pd晶體(111)、(200)、(220)和(311)面心立方結構。NiCo/MWCNT在38.2°處的峰對應于NiOOH的(102)面,而衍射角為81.9°對應的是NiO晶體(222)面的立方晶體結構,因為在堿性環境下Ni和Co較易形成對應的氫氧化物和氧化物。Co的2θ衍射角與Ni的衍射角度相近,因此在圖3中難以看出Co的衍射峰,只有在(e)中能看到2θ為34.0°處的峰對應的是Co晶體的(111)的面心立方結構。
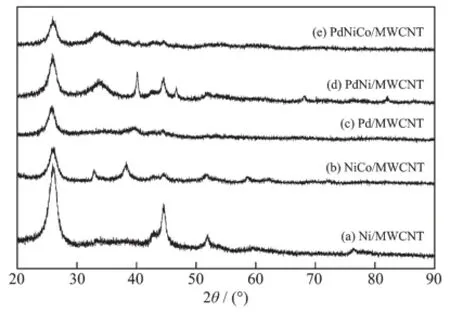
圖3催化劑的XRD圖Fig.3 XRD patterns of the prepared catalysts
圖4為催化劑在1.0 mol·L-1NaOH溶液中的CV圖,從圖中看出,在PdNi/MWCNT和PdNiCo/ MWCNT催化劑上,氫的吸/脫附峰相對于Pd/ MWCNT而言向正向移動。在正向掃描過程中,PdNi/MWCNT和PdNiCo/MWCNT催化劑分別在電位為0.41和0.26 V附近出現明顯的氧化電流,這是催化劑中高價鎳開始形成所引起的;反向掃描過程中,在0.32 V附近出現的還原峰為Ni(Ⅲ)還原到Ni(Ⅱ)的特征峰。此外,PdNiCo/MWCNT催化劑在-0.1 V附近有一個明顯的還原峰,它對應于Ni(Ⅱ)還原到Ni單質的過程,但PdNi/MWCNT上沒有該還原峰,可能與其中鎳的量較少有關(見表1)。從圖4還可看出,Pd/MWCNT,PdNi/MWCNT和PdNiCo/ MWCNT 3種催化劑均在-0.38、-0.35和-0.32 V出現Pd的特征還原峰,對應的峰電流分別為14.2、21.7和26.6mA·cm-2。

圖4催化劑在1.0mol·L-1NaOH溶液中的循環伏安曲線,v=50mV·s-1Fig.4 CVs of the prepared catalysts in 1.0mol·L-1NaOH solution at 50 mV·s-1
圖5為催化劑在0.5mol·L-1乙醇存在下的CV圖。在Pd/MWCNT,PdNi/MWCNT和PdNiCo/MWCNT催化劑上,乙醇氧化的起始電位分別在-0.51、-0.51和-0.48 V附近,峰電流密度分別為77.3、89.6和101.8 mA·cm-2,可以看到,在PdNiCo/MWCNT催化劑上的起始電位稍有提前,且峰電流密度差別很大,說明純鈀催化劑在催化乙醇氧化過程中電極表面的Pd會發生少量毒化,使得其催化性能降低,而加入過渡金屬后催化劑抗毒化的能力增強。根據乙醇在鈀基催化劑上的電氧化機理[23],即:(1)乙醇在催化劑表面形成吸附態乙氧基(CH3COad);(2)吸附態羥基(OHad)的形成;(3)CH3COad和OHad反應形成CH3COO-,以及“雙功能效應”可以解釋這一現象,在乙醇氧化反應開始階段(電位<-0.52 V)電極表面吸附了大量的OHads,在第二種金屬存在下,降低了水活化的電位,因此OHads會在水活化之后脫離電極表面,使Pd的活性位點暴露出來。Liang等人的研究表明,CH3COads和OHads在電極表面和溶液間的轉移是反應的決速步驟[23],因此在Ni和NiCo存在時,乙醇氧化的電流密度比純鈀催化劑的大,而Co的加入改善了催化劑的分散性,Pd的活性位點更多,所以PdNiCo/MWCNT對乙醇氧化的電流密度要大于PdNi/MWCNT。從圖5還能看到,催化劑正向掃描的氧化峰比負向掃描的氧化峰大,3種催化劑的正向氧化峰與負向氧化峰的峰電流之比分別為1.3、1.2、1.3。負向掃描氧化峰電流的大小主要取決于正向掃描氧化之后,形成的產物從電極表面轉移到溶液本體的快慢,比例越大說明產物轉移速率越快,電極表面能放出更多的活性位點供下一循環使用,也表明所制備的催化劑有更好的電催化活性以及更好的去除中間體產物的能力[24]。據報道,碳負載的PdNi合金系列催化劑(金屬碳載量20%)對1 mol·L-1乙醇的氧化電流密度最大為29.16 mA·cm-2[25];用乙二醇還原法制備Pd含量20%的PdNi/MWCNT催化劑,在掃速20 mV·s-1時對乙醇的氧化電流為34 mA·cm-2[26]。表明本文所報道的催化劑的活性要優于相關文獻報道的結果。因此,將Pd納米顆粒原位沉積在Ni和NiCo顆粒的表面,有利于增大Pd納米顆粒的催化活性位點,提高了Pd納米顆粒對乙醇氧化的電催化活性,同時降低了催化劑中的Pd含量,有利于降低燃料電池的成本。

圖5 催化劑在1.0mol·L-1NaOH+0.5mol·L-1EtOH溶液中的循環伏安曲線,v=50mV·s-1Fig.5 CVs of the prepared catalysts in 1.0mol·L-1NaOH+0.5 mol·L-1EtOH solution at 50mV·s-1
利用恒電位階躍比較了不同催化劑的電活性穩定性,圖6為催化劑在電位為-0.4 V時乙醇氧化電流的變化情況。在3種催化劑上,乙醇氧化的電流密度在電解初始階段都迅速下降,這是因為電極/溶液界面雙電層充電,以及界面處乙醇濃度迅速下降引起的。但在約100 s之后電流都趨于穩定。恒電位電解600 s后,在Pd/MWCNT、PdNi/MWCNT和 PdNiCo/MWCNT催化劑上乙醇氧化的電流密度分別為2.7、4.1和8.6mA·cm-2,進一步表明PdNiCo/ MWCNT催化劑對乙醇氧化具有較強的電活性。
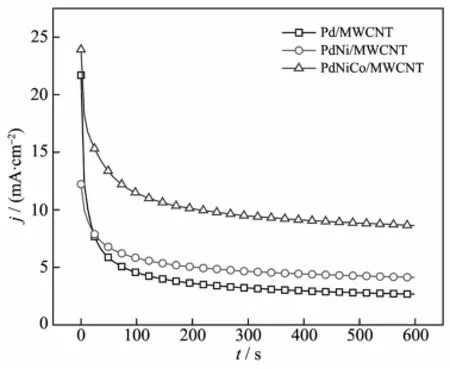
圖6 催化劑在1.0 mol·L-1NaOH+0.5mol·L-1EtOH溶液中的恒電位階躍曲線,階躍電位為-0.4 VFig.6 Chronoamperograms obtained on the prepared catalysts at-0.4 V in 1.0mol·L-1NaOH+0.5 mol·L-1EtOH solution
PdNiCo/MWCNT催化劑在不同乙醇濃度下的循環伏安曲線見圖7。從圖7a可知,隨著乙醇濃度的增大,電流密度也相應地增大,但當濃度達到1.5 mol·L-1時峰電流開始下降,在乙醇濃度0.1~0.7 mol·L-1范圍內,峰電流幾乎呈直線增加,此后增加緩慢(見圖7b)。在半峰電位之前電極表面進行的反應是OHads的形成,催化劑對乙醇氧化的峰電流是OHads和CH3COads共同作用的結果。OHads吸附到電極表面的濃度隨乙醇濃度的增大而增大,在較低濃度下,電極表面的OHads和CH3COads濃度并未達到飽和,且在較低電位時,OHads的吸附與CH3COads的反應是相對獨立進行的,當濃度高于1.5mol·L-1時,隨電位慢慢變大,OHads和CH3COads的濃度也增大了,趨近于達到飽和狀態時在電極表面就會形成競爭關系,在較高電位下,CH3COads的濃度將會占主導地位,CH3COads大量遷移到電極表面并將取代OHads的位置,OHads向電極表面遷移將受限,由此也將導致峰電流的下降[23]。在乙醇濃度為1.5mol·L-1,電位為0.019 V時催化劑對乙醇氧化的峰電流密度為160.6mA·cm-2,當電位超過此峰值電位時,電極表面的Pd開始發生氧化反應,NiCo也相應被氧化而形成各自對應的氧化物或氫氧化物,電極表面Pd的活性表面積減小,阻礙了對乙醇的催化作用而導致電流密度下降。正向掃描形成的氧化物或氫氧化物在反向掃描過程中被還原為金屬,可作為下一個循環的活性物質繼續發揮其催化作用。從圖7c看出,隨乙醇濃度增大,峰電位的增幅也減小,可能是OHads和CH3COads之間的反應達到了一個相對穩定的狀態,導致在較高乙醇濃度時的峰電位增加變緩。
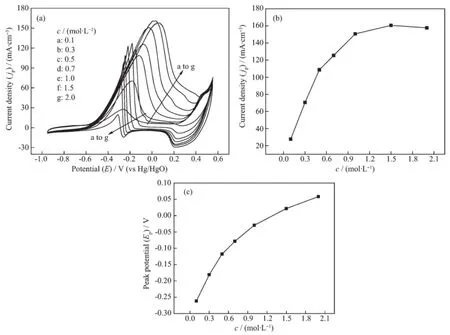
圖7 (a)PdNiCo/MWCNT在1.0mol·L-1NaOH中,不同濃度乙醇(圖中數字)的循環伏安曲線,v=50mV·s-1, (b)乙醇濃度與峰電流的關系,(c)乙醇濃度與峰電位的關系Fig.7 CVs of PdNiCo/MWCNT catalyst in 1.0mol·L-1NaOH with EtOH range from 0.1mol·L-1to 2.0mol·L-1at50mV·s-1(a), plots of EtOH concentration vs anodic peak current density jp(b)and EtOH concentration vs anodic peak potential Ep(c)
不同電位掃描速度下PdNiCo/MWCNT對乙醇氧化的循環伏安圖見圖8a。從圖8b看到乙醇氧化的峰電流在掃速低于100 mV·s-1時呈直線增加,掃速進一步增大時峰電流緩慢增加,主要原因是電極附近乙醇的氧化反應速率隨掃速增大而增大,乙醇濃度下降的趨勢也增強,整個電極反應過程受電極附近乙醇濃度變化影響較大,且本體溶液中的乙醇擴散到電極附近的速率有限,在較高的掃描速度下電極反應是濃差極化控制過程[6]。圖8c表明,峰電位隨掃速增大也呈現正向移動的趨勢,除在低掃速下增幅稍大之外,在50mV·s-1之后,峰電位隨掃速變化呈現緩慢的增加,可能與過電位對OHads的形成以及在電極表面的遷移有較大影響,而對CH3COads的形成和遷移影響不大有關。在反向掃描過程中,Pd的還原電位負移,有可能是Pd的氧化物表面吸附了OHads,OHads的解吸需要在一定的過電位下才能進行。

圖8 PdNiCo/MWCNT在1.0mol·L-1NaOH+0.5mol·L-1EtOH溶液中不同掃速(圖中數字)的循環伏安曲線(a),掃速與峰電流的關系(b),掃速與峰電位的關系(c)Fig.8 CVs of PdNiCo/MWCNT catalyst in 1.0mol L-1NaOH+0.5mol L-1EtOH solution at various sweep rates(a), plots of sweep rate vs the anodic peak currency density jp(b)and sweep rate vs the anodic peak potential Ep(c)
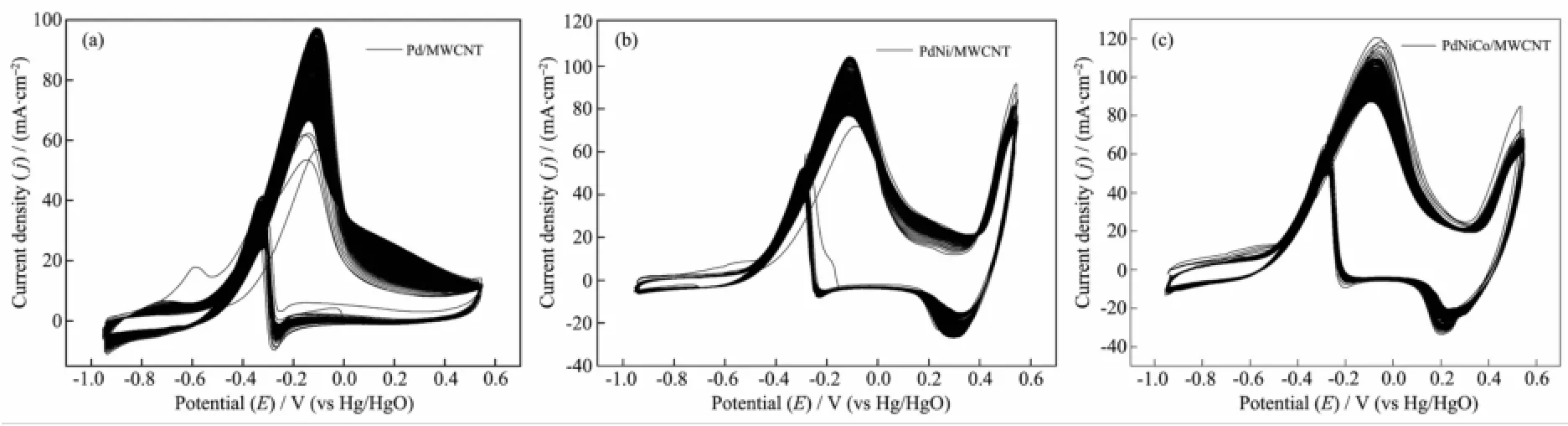
圖9 Pd/MWCNT(a),PdNi/MWCNT(b)和PdNiCo/MWCNT(c)在1.0mol·L-1NaOH+0.5mol·L-1EtOH溶液中,連續循環掃描400次的循環伏安曲線,v=100mV·s-1Fig.9 CVs of Pd/MWCNT(a),PdNi/MWCNT(b)and PdNiCo/MWCNT(c)catalysts in 1.0mol·L-1NaOH+0.5mol·L-1EtOH solution for consecutive 400 cycles at100mV·s-1
采用連續循環伏安法進一步研究了催化劑對乙醇電催化氧化活性的穩定性,圖9為催化劑在含0.5mol·L-1EtOH的1.0 mol·L-1NaOH溶液中,連續進行400次循環掃描的結果,3種催化劑的CV圖形沒有太大變化,隨著掃描次數的增加,除峰電位附近的電流密度有所下降外,其他電位下的電流密度并未發生很大的變動。3種催化劑對應的最大峰電流密度分別為97.4、103.9和112.4mA·cm-2,循環400次之后分別為66.5、76.8和87.7 mA·cm-2,其下降的降幅分別為31.7%、26.1%、22.0%。這是因為隨著反應的進行,溶液中的乙醇濃度有所下降,而在峰電位附近時乙醇濃度對電極反應的影響較大,從而導致峰電流下降比較明顯。對比3種催化劑的CV圖形不難看出,純鈀催化劑的催化性能受乙醇濃度變化更為明顯,峰電流下降更多,這也說明它的穩定性不如摻雜過渡金屬催化劑的穩定性,其穩定性次序為PdNiCo/MWCNT>PdNi/MWCNT>Pd/ MWCNT。
3 結論
以水合肼為還原劑,制備了負載于碳納米管上的納米鎳和納米鎳鈷顆粒。以此顆粒為還原劑與PdCl2溶液反應,還原Pd2+得到負載在多壁碳納米管表面的PdNi/MWCNT和PdNiCo/MWCNT催化劑。采用掃描電鏡,透射電鏡和X射線粉末衍射對所制備的催化劑的形貌和微結構進行了表征,結果顯示Ni/MWCNT和NiCo/MWCNT顆粒是由直徑分別為8~15和8~13 nm的小顆粒團聚形成的,粒徑分別約為250和150 nm,Pd-Ni/MWCNT和PdNiCo/ MWCNT催化劑是由粒徑為8~10和5~8 nm小顆粒形成的、粒徑為60~100和30~50 nm球形顆粒。EDS數據顯示,Pd2+與Ni/MWCNT和NiCo/MWCNT反應以原位沉積的方式進行,形成的Pd納米顆粒包覆在鎳和鎳鈷顆粒表面。采用循環伏安和恒電位階躍技術,對所制備的催化劑在堿性環境中對乙醇的電氧化催化活性進行了測試。結果表明,三金屬對乙醇的電氧化催化活性最高,乙醇氧化的峰電流密度達101.8mA·cm-2,并表現出更好的穩定性。
[1]Rousseau S,Coutanceau C,Lamy C,et al.J.Power Sources, 2006,158(1):18-24
[2]ZhouW J,Zhou ZH,Song SQ,etal.App l.Catal.B:Environ., 2003,46:273-285
[3]Mohammad K,Forouzan A,Ham id R L Z Z,et al.J.Fuel Chem.Technol.,2013,4(1):91-95
[4]TANG Ya-Wen(唐亞文),BAO Jian-Chun(包建春),ZHOU Yi-Ming(周益明),et al.Chinese J.Inorg.Chem.(無機化學學報),2003,19(8):905-908
[5]CHEN Ying(陳瀅),TANG Ya-Wen(唐亞文),GAO Ying(高穎),et al.Chinese J.Inorg.Chem.(無機化學學報),2008, 24(4):560-564
[6]CHEN Qing-Hua(陳清華),YIQing-Feng(易清風).Chinese J.Inorg.Chem.(無機化學學報),2015,31(6):1145-1152
[7]Xu CW,Shen PK,Liu Y L.J.Power Sources,2007,164(2): 527-531
[8]LI Ruo-Shi(李若詩).Thesis for the Doctorate of Fudan University(復旦大學博士論文).2013.
[9]Ding K Q,Yang H W,Cao Y L,et al.Mater.Chem.Phys., 2013,142(1):430-411
[10]Amideddin N,Abbas A K,Yadollah M,et al.Electrochim. Acta,2014,147:192-200
[11]Yang G H,Zhou Y Z,Pan H B,et al.Ultrason.Sonochem., 2016,28:192-198
[12]YIN Wen-Ping(陰文平).Thesis for the Master of Tianjin University(天津大學碩士論文).2010.
[13]Mao H,Huang T,Yu A S.Electrochim.Acta,2015,174:1-7
[14]Zhang Yu(張宇).Thesis for the Master of Shenyang Ligong University(沈陽理工大學碩士論文).2013.
[15]YiQ F,Sun L Z,Liu X P,etal.Fuel,2013,111:88-95
[16]Zhang Z Y,Xin L,Sun K,et al.Int.J.Hydrogen Energy, 2011,36(20):12686-12697
[17]Tsui L,Zafferoni C,Lavacchi A,et al.J.Power Sources,2015,293:815-822
[18]Antolini E,Salgado J R C,Gonzalez E R.J.Electroanal. Chem.,2005,580:145-154
[19]Siracusano S,Stassi A,Baglio V,et al.Electrochim.Acta, 2009,54:4844-4850
[20]Antolini E,Salgado J R C,Gonzalez E R.Appl.Catal.B: Environ.,2006,63:137-149
[21]JIN Chun-Gui(晉傳貴),TAN Jie(檀杰).J.Anhui University of Technology(安徽工業大學學報),2007,24(1):36-38
[22]MarchionniA,BevilacquaM,BianchiniC,etal.ChemSusChem, 2013,6:518-528
[23]Liang Z X,Zhao T S,Xu J B,et al.Electrochim.Acta, 2009,54:2203-2208
[24]Manohara R,Goodenough JB.J.Mater.Chem.,1992,2:875-887
[25]LI Qiao-Xia(李巧霞),LIU Ming-Shuang(劉明爽),MAO Hong-Min(毛宏敏),et al.J.Shanghai University of Electric Power(上海電力學院學報),2012,28(6):565-568
[26]Chen W M,Zhang Y,Wei X F.Int.J.Hydrogen Energy, 2015,40(2):1154-1162
In Situ Formation of Ternary Pd-Ni-Co Nanocatalyst on MWCNT for Ethanol Electro-oxidation in Alkaline M edia
CHEN Qing-Hua1YIQing-Feng*,1,2YANG Zheng1ZHOU Xiu-Lin1LIU Xiao-Ping1NIE Hui-Dong1XUGuo-Rong1
(1School of Chemistry and Chemical Engineering,Hunan University of Science and Technology,Xiangtan,Hunan 411201,China) (2Key Laboratory of Theoretical Organic Chemistry and Function Molecule,Ministry of Education, Hunan University of Science and Technology,Xiangtan,Hunan 411201,China)
Ni/MWCNT and NiCo/MWCNT nanoparticleswere firstly prepared by chemical reductionmethod with hydrazine as reductant in a mixture medium of water and alcohol using Multi-walled carbon nano-tubes (MWCNTs)as the support.PdNi/MWCNT and PdNiCo/MWCNT catalysts were then prepared through an in-situ reduction by the reaction of the Ni/MWCNT and NiCo/MWCNT with the aqueous PdCl2solution respectively. SEM and TEM images show that the large aggregates on the surface of MWCNTs with the diameter of 30~100 nm are composed of small particles with the diameter of 5~10 nm.The ternary PdNiCo catalyst presents the smaller diameter than PdNi catalyst,as well as the better dispersity on MWCNT.ICP and EDS analyses reveal that Pd nanoparticles are directly reduced and coated on the surface of Ni and NiCo nanoparticles.The electrochemical activity of the catalysts towards ethanol oxidation in alkalinemedia has been examined by cyclic voltammetry(CV)and chronoamperometry(CA)methods.PdNiCo/MWCNT catalyst presents an anodic peak current density of 101.8 mA·cm-2for ethanol oxidation inalkalinemedium.Long-term cyclic voltammetric tests show superior stability of Pd nanoparticles upon addition of Niand NiCo to the support.
Pd catalyst;ethanol oxidation;Ni nanoparticles;fuel cell;in situ reduction
O614.82+3
A
1001-4861(2016)07-1161-09
10.11862/CJIC.2016.163
2015-12-17。收修改稿日期:2016-03-30。
國家自然科學基金(No.21376070)和湖南省自然科學基金(No.14JJ2096)資助項目。
*通信聯系人。E-mail:yqfyy2001@hnust.edu.cn;會員登記號:S060019184M。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
