大鼠血漿中非諾貝特活性代謝物含量測定及藥代動力學研究
2016-12-21 06:51:00張媚媚柳曉蕊尹一子
中國藥業 2016年21期
張媚媚,林 蔚,柳曉蕊,尹一子
(廣州醫科大學附屬腫瘤醫院藥學部,廣東 廣州 510095)
大鼠血漿中非諾貝特活性代謝物含量測定及藥代動力學研究
張媚媚,林 蔚,柳曉蕊,尹一子
(廣州醫科大學附屬腫瘤醫院藥學部,廣東 廣州 510095)
目的 建立測定非諾貝特在大鼠血漿中的活性代謝物非諾貝特酸含量的高效液相色譜(HPLC)法。方法 以乙腈沉淀蛋白、酮洛芬為內標,采用Acclaim?120 C18柱,流動相為乙腈-0.2%磷酸溶液(50∶50),檢測波長為297 nm。結果 非諾貝特酸和酮洛芬保留時間為9.1,19.5 min;非諾貝特酸質量濃度在0.02~100.00 g/mL范圍內與峰面積線性關系良好(r=0.999 4);提取回收率在97.74% ~104.13%范圍內,日內和日間精密度的 RSD均小于7%(n=5),穩定性試驗結果良好。結論 自制非諾貝特制劑在大鼠體內的生物利用度遠高于非諾貝特原料藥,為臨床非諾貝特藥物檢測和體內藥代動力學研究提供了有效的技術支持。
非諾貝特酸;高效液相色譜法;血藥濃度
非諾貝特是第2代苯氧芳酸類調血脂藥物,調脂作用確切,是降低三酰甘油(TG)的首選[1]。作為前藥,非諾貝特在體內經酯酶作用下,可迅速代謝成非諾貝特酸而起調血脂作用,具有明顯的降低血清總膽固醇(TC)、TG和升高高密度脂蛋白(HDL)的作用,適用于治療高三酰甘油血癥和高膽固醇血癥,療效優于氯貝特,且不良反應較少[2]。非諾貝特的水溶性非常差,在水中幾乎不溶[3],屬于生物藥劑分類中的第2類藥,溶解度低,滲透性高[4]。口服生物度低,直接影響了其臨床療效[5]。非諾貝特在體內代謝為非諾貝特酸后,在血漿中檢測不到前者的原形[1]。為了解非諾貝特在大鼠體內的藥代動力學過程,本研究中以酮洛芬為內標[6],建立了高效液相色譜(HPLC)法用作非諾貝特在大鼠體內藥代動力學的研究方法,并對比了自制非諾貝特制劑與原料藥在大鼠體內的生物利用度。現報道如下。
1 儀器、試藥與動物
1.1 儀器
UltiMate?3000 Basic Automated型高效液相色譜儀;Chromeleon 6.8色譜數據分析系統(Dionex Corporation Bannockburn IL USA),包括四元梯度泵、在線脫氣機、自動進樣器、柱溫箱(Timberline Instruments,Inc.);Denver TP-4101型電子天平(Denver Instrument);Millipore Milli Q UV Plus型純凈水裝置 (American Instrument Exchange Inc.);Microfuge?高速離心機(Beckman Coulter);VX-100型渦旋混合器(National LabnetCo.,Inc.)。
1.2 試藥
非諾貝特(美國藥典標準,純度為99.8%,香港先進技術工業有限公司);非諾貝特酸標準品(美國Biofine International<Vancouver,BC>公司);酮洛芬標準品(美國Hawkins Pharmaceutical<Minneapolis,MN>公司);乙腈、甲醇(美國FisherScientific<FairLawn,NJ>公司)。空白血漿(Fisher Scientific,Fair Lawn,NJ)取自健康Wistar大鼠,由美國LAMPIRE Biological Laboratories(Pipersville,PA)公司提供。乙腈、甲醇為色譜純,其他試劑為分析純。
1.3 動物
Wistar大鼠,動物實驗符合動物實驗倫理會《實驗動物福利倫理原則》的要求,進行設計并開展,動物合格證編號為SYXK(粵)2010-0104,室溫下單獨飼養。
2 方法與結果
2.1 血藥濃度測定
2.1.1 色譜條件
預柱:Phenomenex C18柱(12.5 mm×4 mm,5 μm);色譜柱:Acclaim?120 column C18柱(150 mm×4.6 mm,2.5 μm);流動相:乙腈-0.2%磷酸溶液(50∶50),用前經過0.5 μm微孔濾膜過濾脫氣;流速:1.0 mL/min;柱溫:30℃;進樣量:20 μL;在線脫氣;檢測波長:297 nm。
2.1.2 溶液配制
非諾貝特酸標準貯備液:經過預試驗,血漿樣品中未出現非諾貝特的色譜峰。精密稱取非諾貝特酸10 mg,置10 mL容量瓶中,用乙腈溶解并稀釋至刻度,搖勻,配成質量溶度為1 g/L的非諾貝特酸母液;將非諾貝特酸母液逐級用乙腈稀釋,得100,5,0.1 μg/mL的標準貯備液,-4℃冷藏備用。
內標溶液:精密稱取酮洛芬標準品 8.0 mg,置100 mL容量瓶中,用乙腈溶解并稀釋至刻度,搖勻,配成質量濃度為80 μg/mL的溶液,-4℃冷藏備用。
2.1.3 血漿樣品處理
取血漿100 μL,加入內標酮洛芬溶液40 μL,混勻,加入乙腈 160 μL,渦旋混合2 min,10 000 r/min離心10 min,取上清液進樣20 μL,記錄色譜圖,見圖1。
2.1.4 方法學考察
專屬性試驗:取空白血漿100 μL,加入乙腈200 μL,按2.1.3項下方法操作,得空白血漿色譜圖(圖1 A);配置含非諾貝特酸和內標的空白血漿溶液,依法處理,得非諾貝特酸和內標提取后的血漿標準品色譜圖(圖1 B)。可見,內標和非諾貝特酸的保留時間分別為9.1 min和19.5 min,互不干擾出峰時間。結果表明,乙腈能夠去除血漿中大部分雜質,不干擾色譜峰。擬訂色譜條件下內標和非諾貝特酸方法專屬性良好,可用于大鼠血漿樣本中非諾貝特酸的測定。色譜圖見圖1。
絕對回收率:取空白血漿適量,制備低、中、高3個質量濃度非諾貝特酸血漿樣品(質量濃度分別為0.05,2.50,80.00 μg/mL)和僅含內標(質量濃度為80 μg/mL)的血漿樣品,按2.1.3項下方法操作,每個質量濃度進行5樣品分析,獲得相應的峰面積為 A1。同時,以流動相代替血漿制備低、中、高3個質量濃度的非諾貝特酸供試品溶液(質量濃度分別為0.05,2.50,80.00 μg/mL)和內標樣品溶液(質量濃度為80.00 μg/mL),每個質量濃度進行5樣品分析,獲得標準溶液色譜峰面積為 A0,以相應濃度的 A1/A0×100比值計算非諾貝特酸和內標的回收率。結果見表1。結果表明,非諾貝特酸低、中、高質量濃度及內標的絕對回收率范圍為99.82%~105.50%,RSD<5%(n=5),表明方法絕對回收率良好。
相對回收率:取空白血漿適量,制備低、中、高3個質量濃度非諾貝特酸血漿樣品(質量濃度分別為0.05,2.50,80.00 μg/mL),按2.1.3項下方法操作,每個質量濃度進行5樣品分析,計算相對回收率,評價方法的準確度。結果表明,非諾貝特酸的平均相對回收率均為99.26%~100.15%,表明方法的準確度良好。結果見表2。
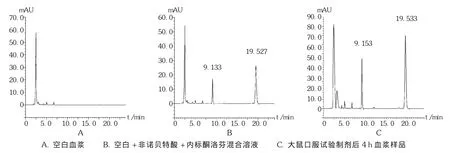
圖1 高效液相色譜圖
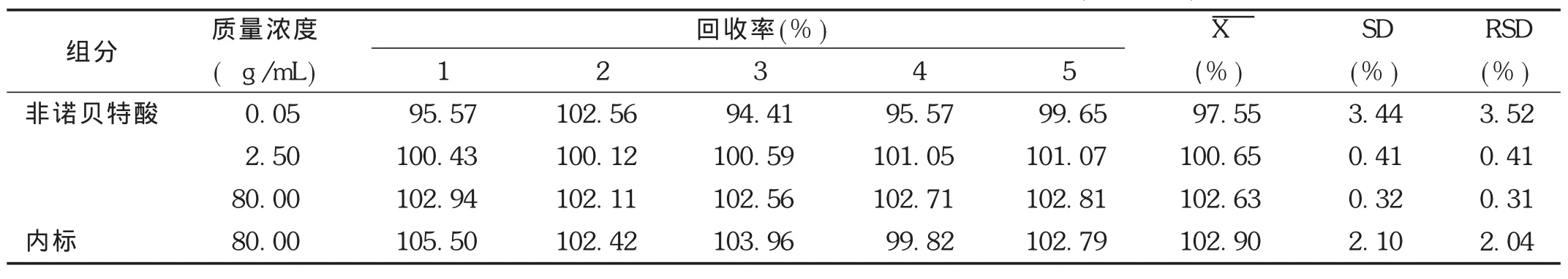
表1 非諾貝特酸和內標在Wistar大鼠血漿中的絕對回收率(%,n=5)
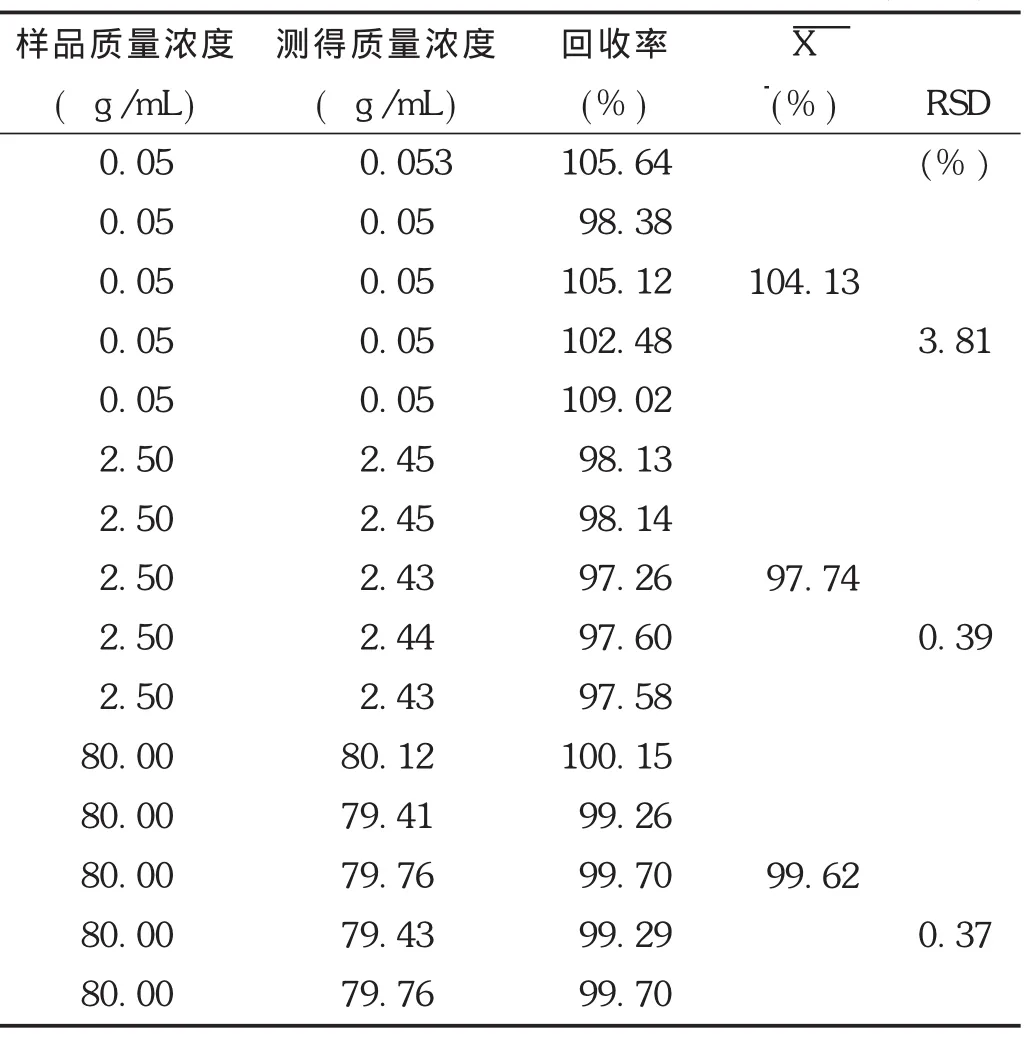
表2 非諾貝特酸在Wistar大鼠血漿中的相對回收率(n=5)
線性關系考察:取非諾貝特酸標準貯備液適量,氮氣吹干,加入空白血漿100 μL,配制成相當于非諾貝特酸血漿質量濃度為0.02,0.05,0.10,1.00,2.50,5.00,20.00,50.00,100.00 μg/mL的系列溶液,按2.1.3項下方法操作,每個質量濃度進樣5次,進樣20 μL,記錄色譜圖。以非諾貝特酸與內標酮洛芬質量濃度比值(X)為橫坐標、非諾貝特酸與內標酮洛芬峰面積比值(Y)為縱坐標,用加權(W=1/c2)最小二乘法進行回歸運算,得直線回歸方程,Y=24.230 34 X+0.001 8,r=0.999 4(n=9)。結果見表 3,表明非諾貝特酸質量濃度在0.02~100.00μg/mL范圍內與峰面積比值線性關系良好。
最低定量限確定:用標準貯備液系列配制成相當于非諾貝特酸血漿質量濃度為0.02 μg/mL的供試品溶液,氮氣吹干,取空白血漿適量,按2.1.3項下方法操作,進行5樣品平行樣本分析,進樣20 μL。根據當天標準曲線計算每個樣本測得質量濃度和相對偏差。結果見表4。結果表明,該方法所測非諾貝特酸的最低定量下限可達0.02 μg/mL,RSD為9.8%(n=5),小于15%。
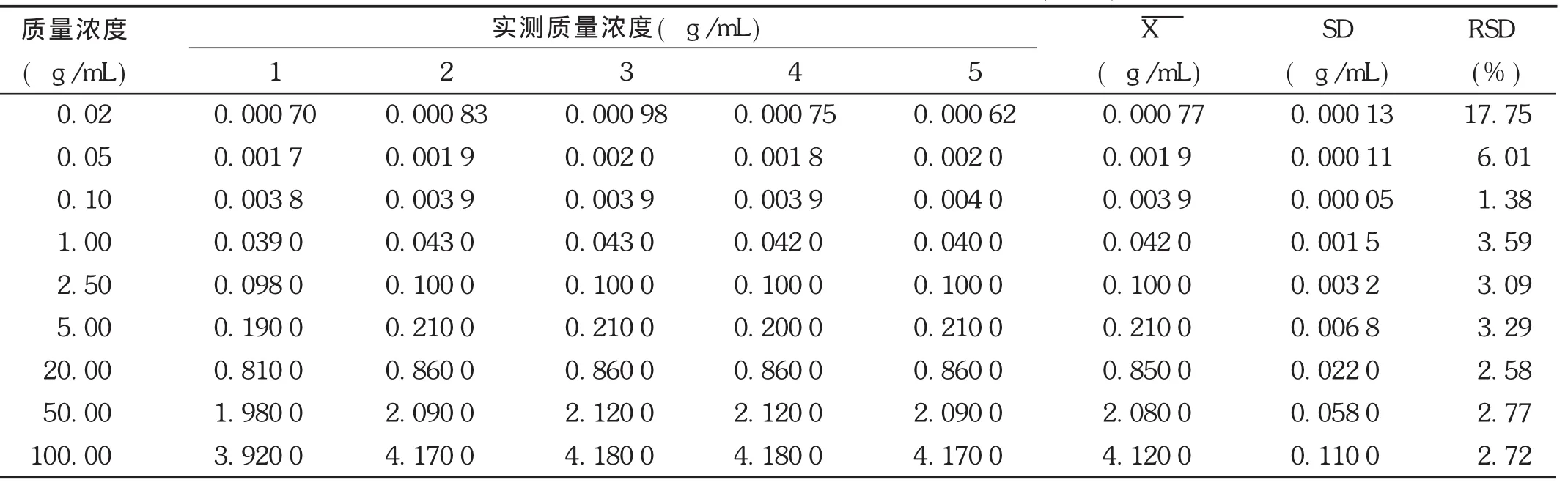
表3 非諾貝特酸在Wistar大鼠血漿中的線性關系(n=5)
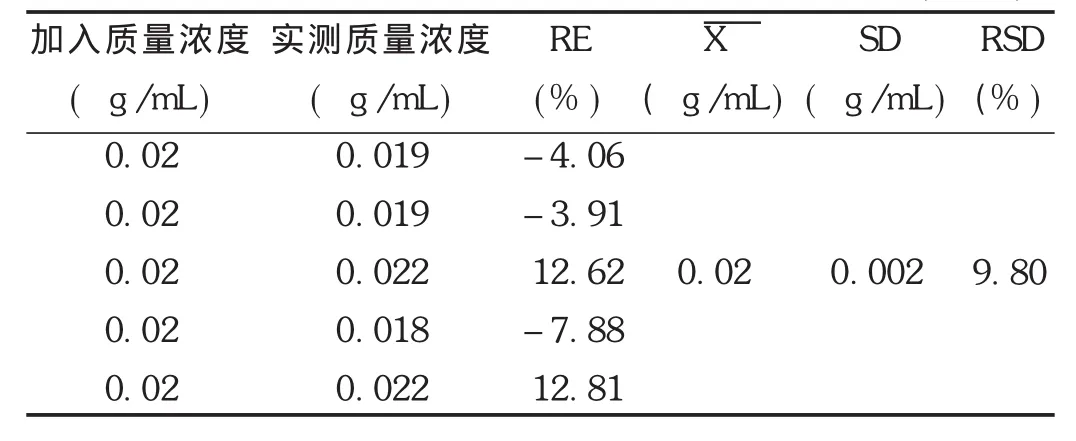
表4 非諾貝特酸在Wistar大鼠血漿中的最低定量限(n=5)
精密度試驗:取空白血漿,按照標準曲線制備項下方法配制低、中、高3個質量濃度(非諾貝特酸質量濃度分別為0.05,2.50,80.00 μg/mL)的樣品,按2.1.3項下方法操作,每個質量濃度進行5樣品平行分析,連續3 d。當日制備當日測定,并根據當日標準曲線,計算樣品測得質量濃度。結果見表5和表6。可見,非諾貝特酸的日內精密度在1.06%~5.67%范圍內(n=5),日間精密度在2.16%~6.87%范圍內,相對誤差均在10%范圍內,表明儀器精密度良好。
穩定性試驗:取空白血漿,按線性關系考察項下方法配制低、中、高3個質量濃度,考察非諾貝特酸樣品提取后室溫放置12 h,-20℃放置1,7,14 d,以及血漿樣品冷凍前、凍融1次、凍融2次的穩定性。結果見表7至表9。可見,非諾貝特酸血漿樣品室溫放置穩定性良好,冷凍1,7,14 d樣品的穩定性良好。凍融2次試驗結果良好,不影響測定結果,符合方法穩定性的要求。
2.1.5 質控樣品含量測定
大鼠血漿樣品分為6批進行測定,每批1條標準曲線,同時分析低、中、高質量濃度的質控樣品(非諾貝特酸質量濃度為0.05,2.50,80.00 μg/mL)并隨行于未知樣品測試匯總。取空白血漿,按照標準曲線制備項下方法配制低、中、高3個質量濃度(非諾貝特酸濃度分別為0.05,2.50,80.00 μg/mL)的質量控制(QC)樣品,每1個質量濃度進行5樣品分析,連續測定3 d,根據當日標準
曲線,計算QC樣品測得質量濃度。根據QC樣品測定結果,計算測定方法的準確度和精密度。根據當批標準曲線求算大鼠血漿樣品質量濃度和QC樣品質量濃度,要求QC樣品質量濃度偏差在±15%范圍內,即回收率控制在85%~115%范圍內,表示儀器檢測正常,否則當批樣品測定結果作廢。結果見表10。QC樣品結果表明,低、中、高QC樣品相對偏差均在±15%范圍內,表明大鼠血漿樣品測定結果可信。

表5 非諾貝特酸在Wistar大鼠血漿中的日內精密度(n=5)
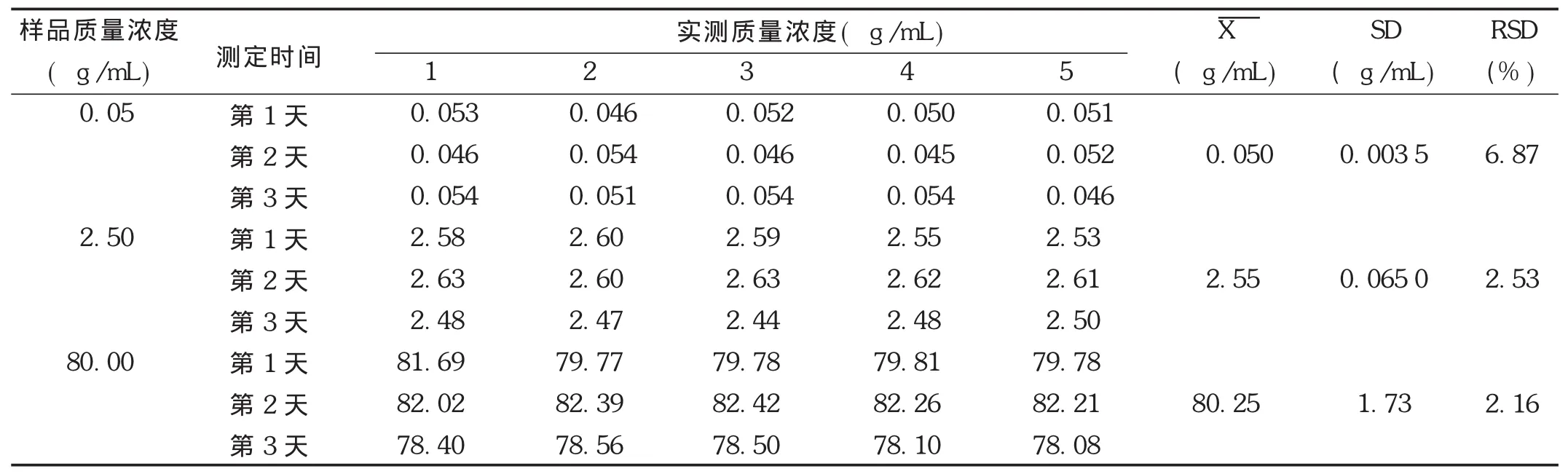
表6 非諾貝特酸在Wistar大鼠血漿中的日間精密度(n=5)
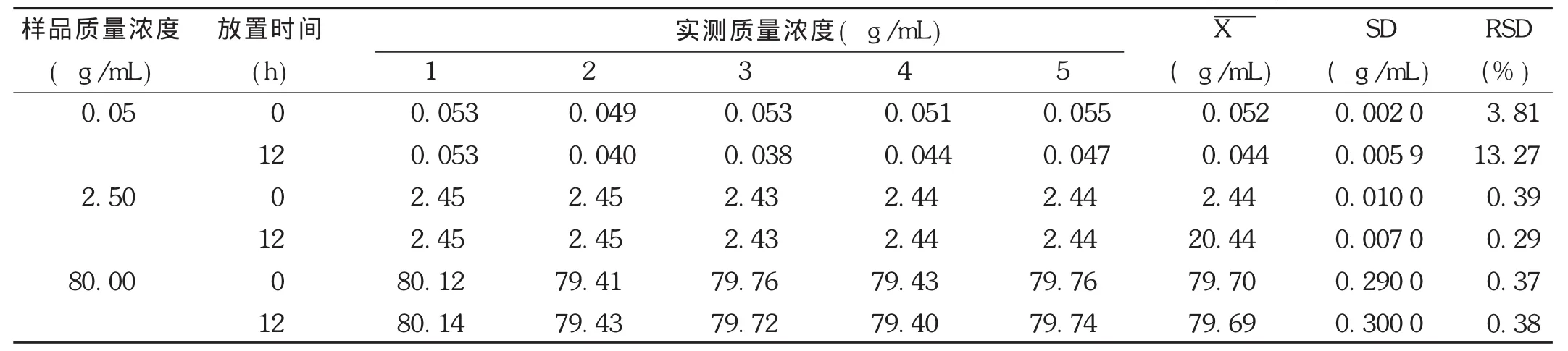
表7 Wistar大鼠血漿中的非諾貝特酸提取后室溫放置12 h的穩定性試驗結果(n=5)
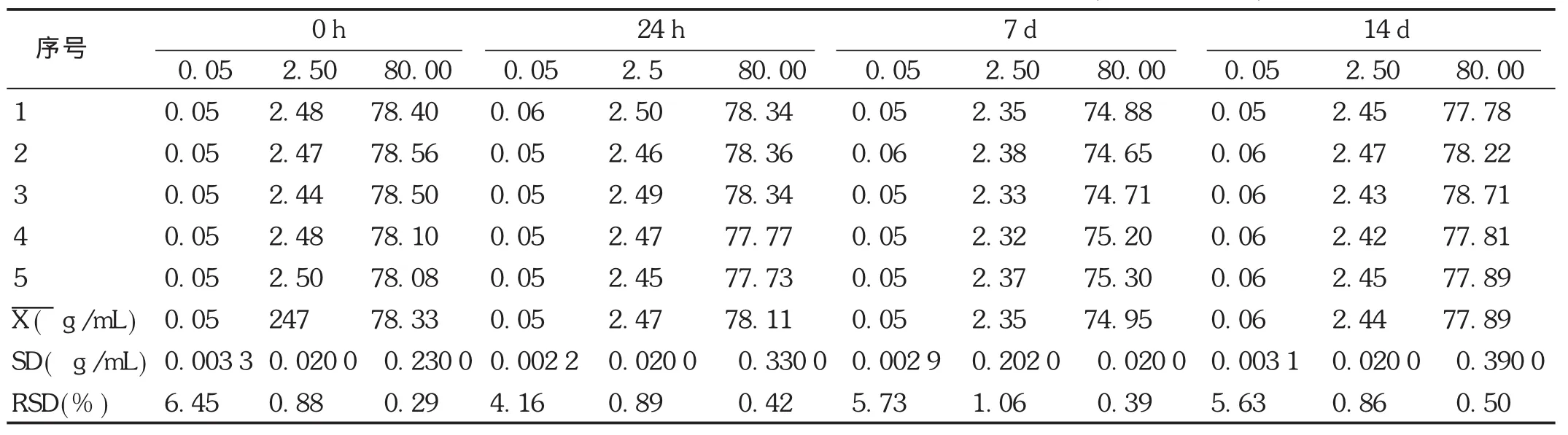
表8 Wistar大鼠血漿中非諾貝特酸的-20℃冷凍穩定性試驗結果(n=5, g/mL)

表9 Wistar大鼠血漿中非諾貝特酸的-80℃凍融穩定性試驗結果(n=5, g/mL)
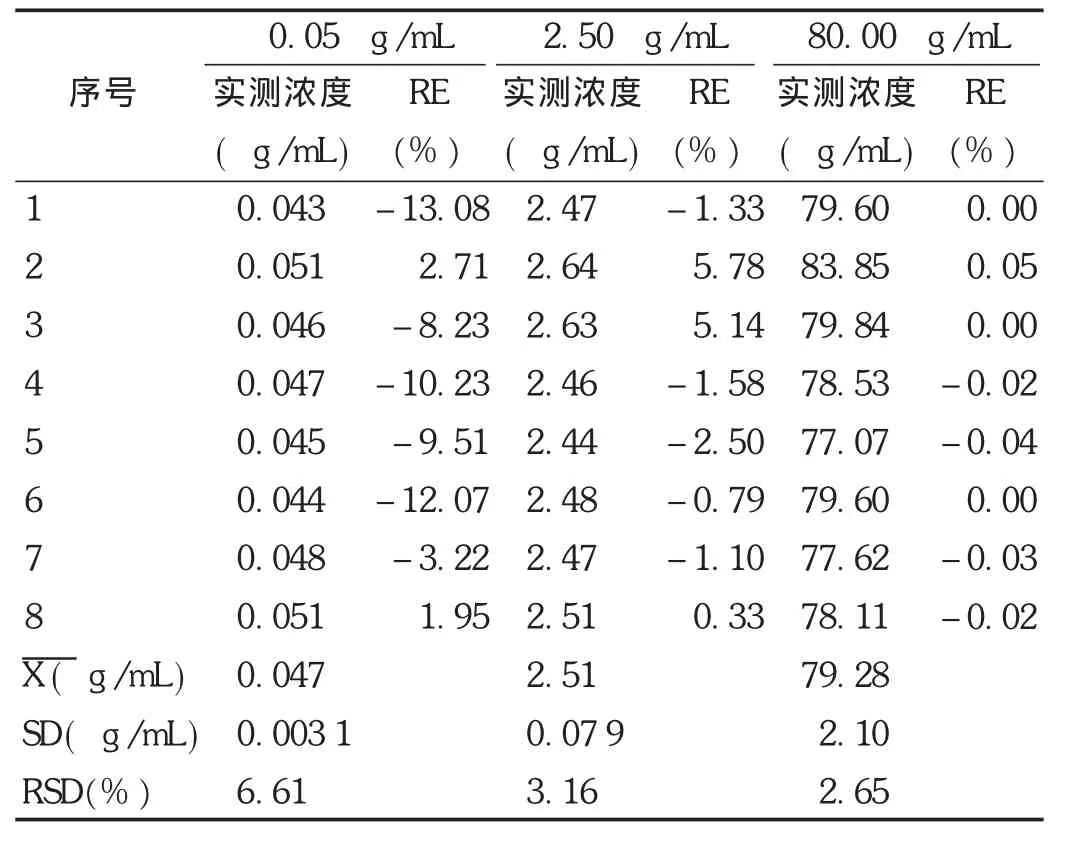
表10 Wistar大鼠血漿中的非諾貝特酸QC樣品含量測定結果
2.2 大鼠單次口服藥代動力學參數研究
2.2.1 大鼠單次口服試驗
預先頸部插管雄性 Wistar大鼠 12只(體質量300~350 g),試驗前3 d可自由飲食。在禁食12 h后,給藥前稱重標號,分為兩組。分別向胃內灌注非諾貝特原料藥、非諾貝特自制制劑混懸劑各 2 mL(溶劑為0.3%的CMC-Na溶液),給藥量為27 mg/kg。分別于灌胃0.25,0.5,0.75,1,2,4,6,8,12,24,36,48 h后,從大鼠的頸部預插管取血0.3 mL,置肝素化離心管中,立即注射37℃保溫的0.3 mL生理鹽水和0.1 mL肝素。所取血樣離心5 min(4 000 r/min),吸取上層血漿,置-20℃冰箱中保存,待測。按2.1.3項下方法制備血漿樣品,采用HPLC法測定。
2.2.2 數據處理
藥代動力學參數計算:根據所測非諾貝特酸血藥濃度-時間數據,利用Winolin非房室模型計算主要藥代動力學參數,包括半衰期(t1/2)、48 h血藥濃度曲線下面積(AUC0-48)、平均滯留時間(MTR)、最大血藥濃度(Cmax)和達峰時間(Tmax)。Cmax和 Tmax采用實測值,AUC為統計矩計算值。
相對生物利用度計算:非諾貝特酸相對生物利用度=AUC0-48(試驗制劑)/AUC0-48(參比制劑)×100%。采取五樣本平行測定,方法學標準偏差(SD)和相對標準偏差(RSD%)采用Excel軟件計算結果。
2.2.3 測定結果與分析
大鼠體內藥代動力學參數表明,非諾貝特的原料藥吸收非常緩慢,口服生物利用度非常低,但自制制劑生物利用度得到明顯提高。相同給藥情況下,非諾貝特自制制劑 Tmax為1.42 h,達峰質量濃度為(58.17± 14.43)μg/mL,吸收速率大于原料藥,血藥濃度曲線下面積比為原料藥的38倍(自制制劑 AUC0-48/原料藥AUC0-48)。詳見圖2和表11。證明自制制劑對其口服吸收具有非常重要的改善作用,提高了難溶性藥物非諾貝特的溶解度和吸收。
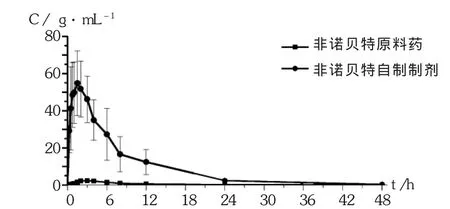
圖2 Wistar大鼠單次口服非諾貝特原料藥及自制制劑的混懸液后的平均血漿濃度-時間曲線(給藥量27 mg/kg,n=6)
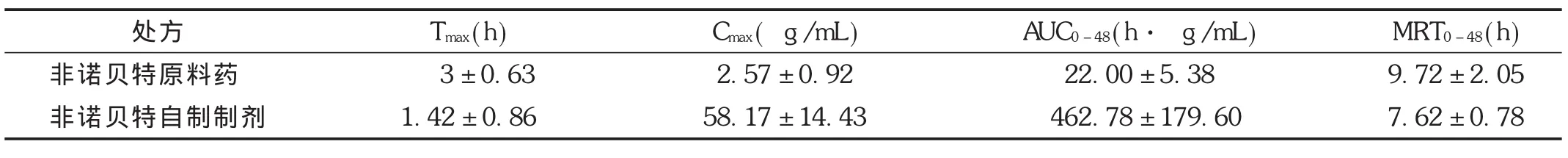
表11 Wistar大鼠單次口服非諾貝特原料藥及自制制劑混懸液的動力學參數(給藥量27 mg/kg,n=6)
3 討論
調脂藥非諾貝特口服后在胃腸道迅速吸收分布,分解代謝為活性代謝產物非諾貝特酸,轉化為非活性代謝物后通過腎臟和糞便排除[7]。因此根據《化學藥物非臨床藥代動力學研究技術指導原則》,并在文獻[8]報道的基礎上進行了改進,建立了非諾貝特酸在大鼠血漿中測定的HPLC法。文獻[9-10]報道了氣相色譜-質譜法、HPLC法[11-12]等用于非諾貝特口服后體內非諾貝特酸藥代動力學的研究,但分離測定都較煩瑣。本研究中采用HPLC法操作快速、簡便,干擾少,影響因素少,分離度良好,靈敏度高,適合藥代動力學或者毒代動力學研究中大量的樣本分析。
文獻[13]報道中建立了3倍量甲醇沉淀蛋白,高速離心后取上清液進樣的方法,結果提取回收率為97.18% ~107.28%。本研究中采取了乙腈沉淀蛋白法提取,以血漿的2倍體積即可達到蛋白完全沉淀的效果,血樣預處理的方法簡便快捷,選擇性好,提取率高,分析的效率高,方法絕對回收率為99.82%~105.50%,RSD<5%,且檢出限低,測定過程穩定,分析費用低。本研究中,采用乙腈 ∶0.2%磷酸溶液(50∶50)為流動相系統,所得的樣品保留時間合適,峰形對稱,色譜分離度和重復性好,避免了流動相中加入緩沖鹽殘留在進樣閥中對高效液相色譜泵、進樣器和色譜柱的損傷,有利于樣本快速、準確地檢測。檢測波長選擇286 nm,為非諾貝特酸的最大吸收波長,靈敏度高,最低定量限達0.02 μg/mL,進樣體積小[14]。選取了酮洛芬為內標,其溶解的特性及色譜行為與非諾貝特酸相似,具有合適的保留時間,提取回收率相近,血漿雜質對其無干擾,與非諾貝特酸峰之間無干擾,性質穩定,價廉易得,滿足作為非諾貝特酸內標的要求。在方法學研究中分別對非諾貝特酸從樣品采集、放置和配制等方面進行了穩定性研究,發現非諾貝特酸在樣品提取后室溫放置12 h,-20℃放置1,7,14 d,以及血漿樣品冷凍前、凍融1次、凍融2次的穩定性均良好。
非諾貝特自制制劑大鼠灌胃給藥后,其藥代動力學參數 Tmax是原料藥的0.5倍,而 Cmax是原料藥的19倍,生物利用度是38倍,表明非諾貝特自制制劑在體內的吸收速度較原料藥明顯加快,吸收程度顯著增加[15]。藥-時曲線符合口服吸收的二室模型[16-17]。本研究中建立的HPLC法,成功地運用于檢測大鼠血漿中非諾貝特的活性代謝產物非諾貝特酸,滿足藥代動力學研究的要求。在給藥劑量范圍內,非諾貝特酸在大鼠體內的藥代動力學符合線性藥代動力學規律。
綜上所述,本研究中采用了比較簡單的色譜條件檢測了非諾貝特在大鼠血漿中的活性代謝產物非諾貝特酸的質量濃度,可及時、準確地計算非諾貝特的代謝率,為臨床非諾貝特藥物檢測和體內藥代動力學研究提供了有效的技術支持。
[1]劉秀美,王 凌,蔣學華.HPLC測定大鼠血漿中非諾貝特的活性代謝物及其藥動學研究[J].華西藥學雜志,2012,27(4): 424-427.
[2]Rosenson RS,Helenowski IB,Tangney CC.Heterogeneous Postprandial Lipoprotein Responses in the Metabolic Syndrome,and Response to Fenofibrate Therapy[J].Cardiovasc Drugs Ther,2010,24(5-6):439-447.
[3]鄭 楊,張志麗,王立紅,等.非諾貝特固體分散體制備工藝研究及比較[J].中國藥劑學雜志(網絡版),2012,10(2): 26-34.
[4]陳桂江.PEG化脂質納米粒促進非諾貝特口服吸收的研究[D].廣州:暨南大學,2015.
[5]陶小妹,孫路路,王淑梅,等.非諾貝特PEG_(2000)-DSPE膠束的制備及在大鼠體內的口服藥動學研究[J].中國藥師,2016,19(4):634-638.
[6]Zhang M,Li H,Lang B,et al.Formulation and delivery of improved amorphous fenofibrate solid dispersions prepared by thin film freezing[J].European journal of pharmaceutics and biopharmaceutics:official journal of Arbeitsgemeinschaft fur Pharmazeutische Verfahrenstechnik eV,2012,82(3):534-544.
[7]李 芳,俸靈林,閻 超.非諾貝特制劑研究進展[J].世界臨床藥物,2011,32(9):552-557.
[8]沈 敏,沈 騰,李中東,等.非諾貝特與代謝物的HPLC測定及在大鼠小腸淋巴轉運中的應用[J].中國藥學雜志,2011,46(9):686-690.
[9]張 丹,劉 曼,王曉琳,等.LC-MS/MS法測定人血漿中非
諾貝特酸[J].國際檢驗醫學雜志,2013,34(13):1 688-1 690.
[10]馬 飛,邢春宇,吳卓娜,等.HPLC-MS/MS測定比格犬血
漿中非諾貝酸濃度及其藥代動力學研究[J].解放軍藥學學報,2013,29(3):196-199.
[11]趙燕燕,白 潔,蘇 芳,等.高效液相色譜法同時測定體內辛伐他汀和非諾貝特酸的含量[J].中國醫院藥學雜志,2011,31(12):998-1 001.
[12]張 瑋,劉守信.非諾貝特膠囊在健康人體內的藥物代謝
動力學研究[J].中國現代醫學雜志,2014,24(31):56-59.
[13]徐 帆,馮恩富,余 .HPLC法測定血漿中非諾貝特活性代
謝物非諾貝酸的濃度[J].中國藥師,2007,10(6):530-532.
[14]馮沁心,黃琴琴,王永祿,等.非諾貝特納米混懸劑大鼠體
內藥動學及其與在體腸吸收動力學相關性[J].中國新藥與臨床雜志,2013,32(9):738-743.
[15]Zhu T,Aasquer JC,Kelly MT,et al.Comparison of the gastrointestinal absorption and bioavailability of fenofibrate and fenofibric acid in humans[J].Journal of Clinical Pharmacology,2010,50(8):914-921.
[16]Buch P,Languth P,Kataoka M,et al.IVIVC in Oral Absorption for Fenofibrate Immediate Release Tablets Using a Dissolution/Permeation System[J].Journal of Pharmaceutical Sciences,2009,98(6):2 001-2 009.
[17]費偉東,吳 超,邱 陽,等.介孔二氧化硅納米粒增溶型非諾貝特片劑的制備及體內外研究[J].中國現代應用藥學,2015,32(9):1 097-1 102.
Determination and Pharmacokinetic Study of Fenofibrate Active Metabolite in Rat Plasma by HPLC
Zhang Meimei,Lin Wei,Liu Xiaorui,Yin Yizi
(Affiliated Cancer Hospital of Guangzhou Medical University,Guangzhou,Guangdong,China 510095)
Objective To develop an HPLC method for determining the plasma drug concentration of fenofibrate active metabolitefenofibric acid.Methods Acetonitrile precipitated protei and ketoprofen were used as the internal standard.The column was Acclaim? 120 C18.The mobile phase consisted of acetonitrile-0.2% phosphoric acid solution(50∶50).The detection wavelength was 297 nm.Results The retention time of fenofibric acid and ketoprofen were about 9.1 min and 19.5 min,respectively.The standard curve was linear in the concentration range 0.02-100.00 μg/mL(r=0.999 4).The recoveries were 97.74% -104.13%.The RSD of intraand inter-day assays were both lower than 7%(n=5).Pretreated solution of fenofibric acid in human plasma was stable.Conclusion The bioavailability of self-made fenofibrate in rats is much higher than that of fenofibrate,which provides effective technical support for the clinical detection of fenofibrate and in vivo pharmacokinetics.
fenofibric acid;HPLC;plasma drug concentration
R965;R969.1;R972+.6
A
1006-4931(2016)21-0028-06
張媚媚(1984-),女,博士研究生,主管藥師,研究方向為緩控釋制劑與靶向制劑,(電話)020-66673666(電子信箱)zhmm03@hotmail.com;尹一子(1962-),女,碩士研究生,主任藥師,研究方向為抗腫瘤藥物的臨床合理應用及個體化給藥方案的設計,本文通訊作者,(電話)020-66673666(電子信箱)yinyizi6262@aliyun.com。
2016-07-13;
2016-08-22)
廣州醫科大學博士/出國留學啟動基金,項目編號:2013C66。
猜你喜歡
現代臨床醫學(2022年4期)2022-09-29 07:38:00
中學生數理化·八年級物理人教版(2021年12期)2021-12-31 03:23:08
昆明醫科大學學報(2021年4期)2021-07-23 01:21:50
中學生數理化·中考版(2020年10期)2020-11-27 01:59:48
中國生殖健康(2019年2期)2019-08-23 08:12:08
云南醫藥(2019年3期)2019-07-25 07:25:14
產品可靠性報告(2017年7期)2017-09-05 09:49:12
海南醫學(2016年8期)2016-06-08 05:43:00
汽車觀察(2016年3期)2016-02-28 13:16:26
醫學研究雜志(2015年9期)2015-07-01 17:28:15
