黃芩高通量轉錄組測序數據組裝和分析
2017-01-06 12:20:57王德富成媛媛牛二波牛顏冰
山西農業科學 2016年8期
王德富,成媛媛,楊 鋒,牛二波,牛顏冰
(山西農業大學生命科學學院,山西太谷030801)
黃芩高通量轉錄組測序數據組裝和分析
王德富,成媛媛,楊 鋒,牛二波,牛顏冰
(山西農業大學生命科學學院,山西太谷030801)
采用高通量測序技術對黃芩的轉錄組進行測序,獲得了29 099 899條reads數據,拼接后得到53 353條Unigene;將所獲得的Unigene與COG,GO,KEGG,Swiss-Prot,NR這5個公共數據庫進行比對,結果發現,分別有10 756,21 950,8 101,20 339,29 288條Unigene可比對到以上5個數據庫中;已注釋的Unigene與COG數據庫比對后按功能可分為25類;根據GO功能可分為三大類57個亞類;經過與KEGG數據庫比對后按照代謝通路可分為116類;利用GetORF軟件進行ORF預測,獲得長度大于300 nt的ORF共20 552個;通過SSR分析,共獲得5 658個SSR標記。獲得的轉錄組信息可為今后進行黃芩分子標記的開發和關鍵基因的克隆及功能分析等研究提供基礎數據。
黃芩;轉錄組測序;生物信息學分析
黃芩(Scutellaria baicalensis Georgi)為唇形科黃芩屬植物的干燥根[1-3],具有清熱燥濕、涼血安胎、解毒等功效,常用于治療胸悶嘔惡、濕熱痞滿、瀉痢、胎動不安等癥[4-6],臨床療效極為廣泛。隨著黃芩在藥物開發中的廣泛應用,其需求量逐年上升,為了滿足日益膨脹的市場需求和減輕對黃芩野生資源的掠奪式采挖壓力,采用生物技術大規模合成有效成分,已成為藥物生產和新藥開發的重要途徑。但由于黃芩目前還未進行全基因組測序,相關基因資料不全,導致黃芩中有效成分生物合成途徑方面的研究比較滯后。而高通量轉錄組測序技術的出現,為研究黃芩活性成分生物代謝途徑提供了重要的基因資源,并為開展黃芩功能基因組學研究提供了全新的思路和方法[7-8]。
轉錄組分析可在不知道物種基因信息的情況下,直接從RNA水平分析細胞代謝規律,揭示基因表達與一些生命現象之間的關系,進而對細胞進行修飾和改造[9-11]。該法特別適合基因信息十分匱乏的物種[12-14],并已在人類[15]、酵母[16]、玉米[17]、擬南芥[18]、大豆[19]、蝴蝶[20]等多個物種中得到廣泛應用。在中藥材方面,郝大程等[21]、李鐵柱等[22]、李瀅等[23]、郭溆[24]分別對虎杖根、杜仲果實和葉片、丹參根、鐵皮石斛和人參進行了轉錄組測序,并發現了一些參與藥效成分生物合成相關的轉錄本和關鍵酶序列。這些研究結果說明,RNA-Seq技術在藥用植物研究中也得到了極其廣泛的應用。
本研究利用Illumina HiSeqTM2500對黃芩進行轉錄組測序和分析,從黃芩轉錄組中識別出與黃芩苷生物合成相關的關鍵基因,并發現了大量的簡單重復序列(SSR)。這些重要基因和SSR序列的發現,為進一步克隆其全長、研究其功能和進行分子標記的開發提供了基礎數據,同時也為藥效成分的生物合成研究奠定了基礎。
1 材料和方法
1.1 材料
2014年8月在山西絳縣2年生黃芩種植基地,分別采集最新長出的黃芩根、莖、葉和花,每個組織選取10株取樣,用蒸餾水沖洗干凈后,置于液氮中冷凍保存,備用。
1.2 方法
1.2.1 總RNA提取及檢測 使用TRIzol(Invitrogen)法提取黃芩4種不同組織的總RNA,并測定其質量。若RIN(RNA完整值)大于8.5,28S/18S大于2,OD260/OD280在1.8~2.2,OD260/OD230大于1.8,則樣品合格,然后將質量較好的RNA樣本等比例混合后進行轉錄組測序。
1.2.2 黃芩cDNA文庫的建立和測序 cDNA文庫構建和轉錄組測序工作委托北京百邁客生物科技有限公司完成。
1.2.3 黃芩轉錄組數據處理和組裝 將測序得到的原始圖像數據轉化為原始讀序(rawreads),去除其中的接頭序列和低質量的reads,即可獲得高質量的干凈讀序(clean reads)。利用Trinity[25]軟件對clean reads進行de novo組裝。
1.2.4 功能注釋和分類 首先通過BLASTX將得到的Unigene分別與核酸和蛋白數據庫進行比對(E-value<10-5),獲得最佳注釋信息;接著用Blast2GO和WEGO軟件對所有GO注釋信息進行GO功能分類統計[26];用COG數據庫進行Unigene功能注釋和分類,了解基因功能分布特性;用KEGG數據庫對Unigene進行功能注釋,分析某個Unigene在細胞代謝通路中的功能定位[27]。
1.2.5 ORF預測與SSR位點搜索 利用GetORF軟件對Unigene進行ORF(Open ReadingFrame)預測,選取預測結果中最長的序列作為該Unigene最終的ORF。使用MISA(http://pgrc.ipk-gatersleben. de/misa/misa.htmlt/)軟件對篩選得到的1 kb以上的Unigenes做SSR序列分析,檢測Unigene序列中SSR的分布特征。
2 結果與分析
2.1 RNA質量檢測
利用Agilent 2100生物分析儀(Agilent Technology,USA)和NanoDrop分光光度計(Thermo,USA)對提取的黃芩RNA進行檢測,結果表明,其RIN值大于8.5,OD260/OD280在1.8~2.2,OD260/OD230大于1.8,28S/18S大于2,說明所得到的RNA質量較好,能夠滿足后續的轉錄組測序試驗(表1)。

表1 RNA質量檢測結果
2.2 黃芩測序數據質量評價及序列組裝分析
經HiSeqTM2500高通量測序獲得29 099 899條讀序(clean data),總核苷酸數達7 326 522 924個,數據量為7.33 G,GC含量為45.87%,Q30(測序錯誤率<1%)值為90.743%(圖1-a);同時堿基分布分析表明,除了由于在read 5′端前十幾個堿基存在明顯的偏向性而導致前端波動較大外,每個測序循環中AT和GC的含量在整個測序過程中基本呈水平線,含量穩定不變(圖1-b),說明所得到的測序數據質量較好,可進行后續分析。
利用Trinity軟件對上述獲得的clean reads進行組裝,得到4538775條contigs,平均長149.51 bp,N50為148 bp;然后再通過contigs之間的相似性以及雙末端信息將其組裝得到107 533條transcripts,平均長2 038 bp,N50為1 257.69 bp;進一步的去冗組裝獲得53 353條Unigenes,平均長1 467 bp,N50為797.64 bp(表2)。所得到transcript和Unigene的N50分別達到1 257.69,797.64 bp,表明組裝片段的完整性比較高。
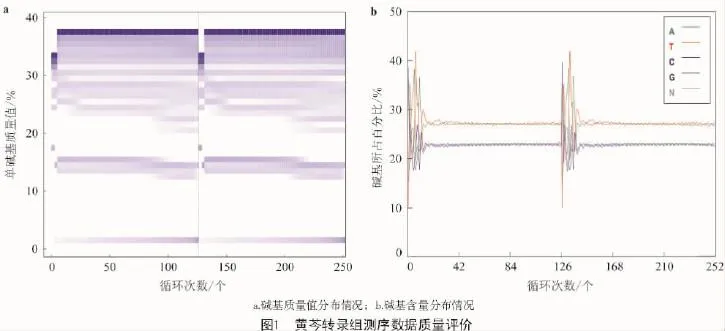
表2 黃芩轉錄組數據組裝統計結果
2.3 Unigene的功能注釋
表3 Unigenes在5個數據庫中的分布情況
將組裝得到的53 353條Unigene與已知的COG,GO,KEGG,Swiss-Prot和NR這5個公共數據庫進行序列比對。從表3可以看出,Unigene在5個不同數據庫中的同源比對數目不同,分別為10 756,21 950,8 101,20 339,29 288條。總共所獲得的同源比對信息數量為29 382條,占Unigene總數量的55.07%,但仍有23 971條(44.93%)序列定位不清楚,這可能與公共數據庫中沒有黃芩全基因組序列有關。
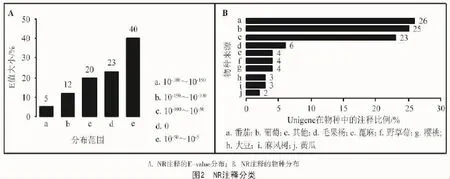
從圖2-A可以看出,比對到NR數據庫中的29 288條Unigenes,有40%的E值分布在10-50~10-5,20%位于 10-100~10-50,12%位于 10-150~10-100,小于10-150的占5%。從匹配的物種來源分析(圖2-B),發現注釋到番茄中Unigene占26%,注釋到葡萄中Unigene占25%,注釋到其他物種中的Unigene相對較少,如注釋到大豆和麻風樹中的Unigene均為3%,蓖麻、野草莓和櫻桃的Unigene均為4%,毛果楊為6%,黃瓜為2%,其余23%注釋到其他物種。
2.4 Unigene的功能分類
由表3可知,有21 950條Unigene(41.14%)獲得GO數據庫注釋。這些Unigene被分在上述3大類的57個亞類中(圖3),其中,生物過程分為25個亞類,獲得的注釋信息最多,共有65749條Unigene,占全部注釋信息的44.33%,這里涉及代謝過程、細胞過程和對刺激的應激效應的Unigene比較多,分別有14 564,13 416,6 291條;其次是細胞成分,分為16個亞類,共有57 243條Unigene,占全部注釋信息的38.60%,涉及到細胞部分、細胞和細胞器的Unigene比較多,分別有13 930,13 802,11 606條;分子功能的注釋信息最少,共有25 315條Unigene,占17.07%,這其中的催化活性與結合排在前2位,分別有11 123,10 106條Unigene。
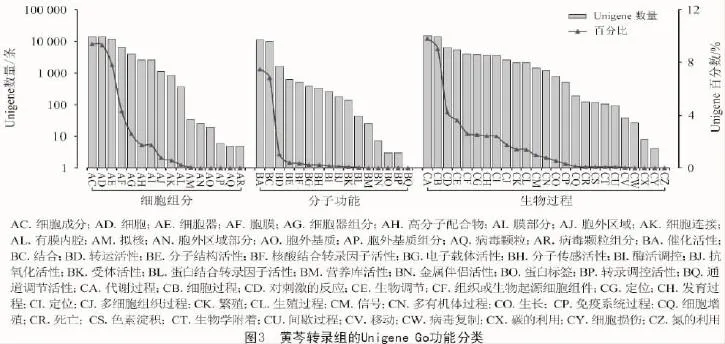
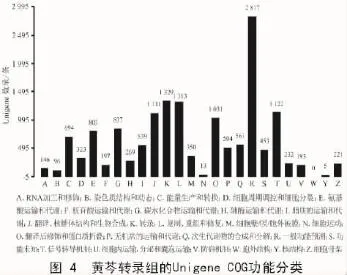
另外,對獲取的Unigene進行了COG數據庫功能注釋,并歸入不同的分類中,共有15 159條Unigene歸入了25種COG分類中(圖4)。其中,一般功能預測類包含Unigene最多(2 817條),其次是轉錄(1 329條)、復制、重組和修復(1 313條)、信號轉導機制(1 122條)、翻譯、核糖體結構和生物合成(1 111條);涉及細胞活性和細胞核結構的Unigene較少,分別僅有13,5條;沒有發現細胞外結構的功能基因。另外,有453條Unigene未被注釋上,該注釋與其他物種注釋結果比較相似。
2.5 Unigene代謝途徑分析
將所獲得的53 353條Unigene映射到KEGG數據庫中,有8 101條(15.18%)Unigene得到注釋,并歸入116種KEGG代謝途徑中。從表4可看出,涉及核糖體代謝途徑的Unigene數量最多(410條/ 5.06%),其次是氧化磷酸化(287條/3.54%)、內質網蛋白加工(270條/3.33%)、剪接體(252條/3.11%)和嘌呤代謝(245條/3.02%)等代謝通路。但歸入生物素代謝和油菜素內酯合成等中的Unigene較少,數量不超過10條。
2.6 ORF與SSR位點分析
為進一步解析黃芩轉錄組編碼的基因信息,利用GetORF軟件預測了已得到注釋的Unigene,共得到53 028個ORF,其中,長度大于300 nt的Unigene最多,占38.81%(20 552條)(圖5)。另外,本研究還利用MISA軟件找出5 658個SSR標記位點,其中,雙堿基型重復最為豐富(2 928個/51.75%),其他類型依次為:單堿基型(1 540個/27.22%)、三堿基型(1 127個/19.92%)、四堿基型(37個/0.65%),五堿基型和六堿基型分別為11,15個,各占0.19%和0.27%(表5)。黃芩SSR位點的分析,將為黃芩的遺傳標記研究提供非常重要的物質資源和依據。

表4 黃芩轉錄組Unigene KEGG代謝途徑分類
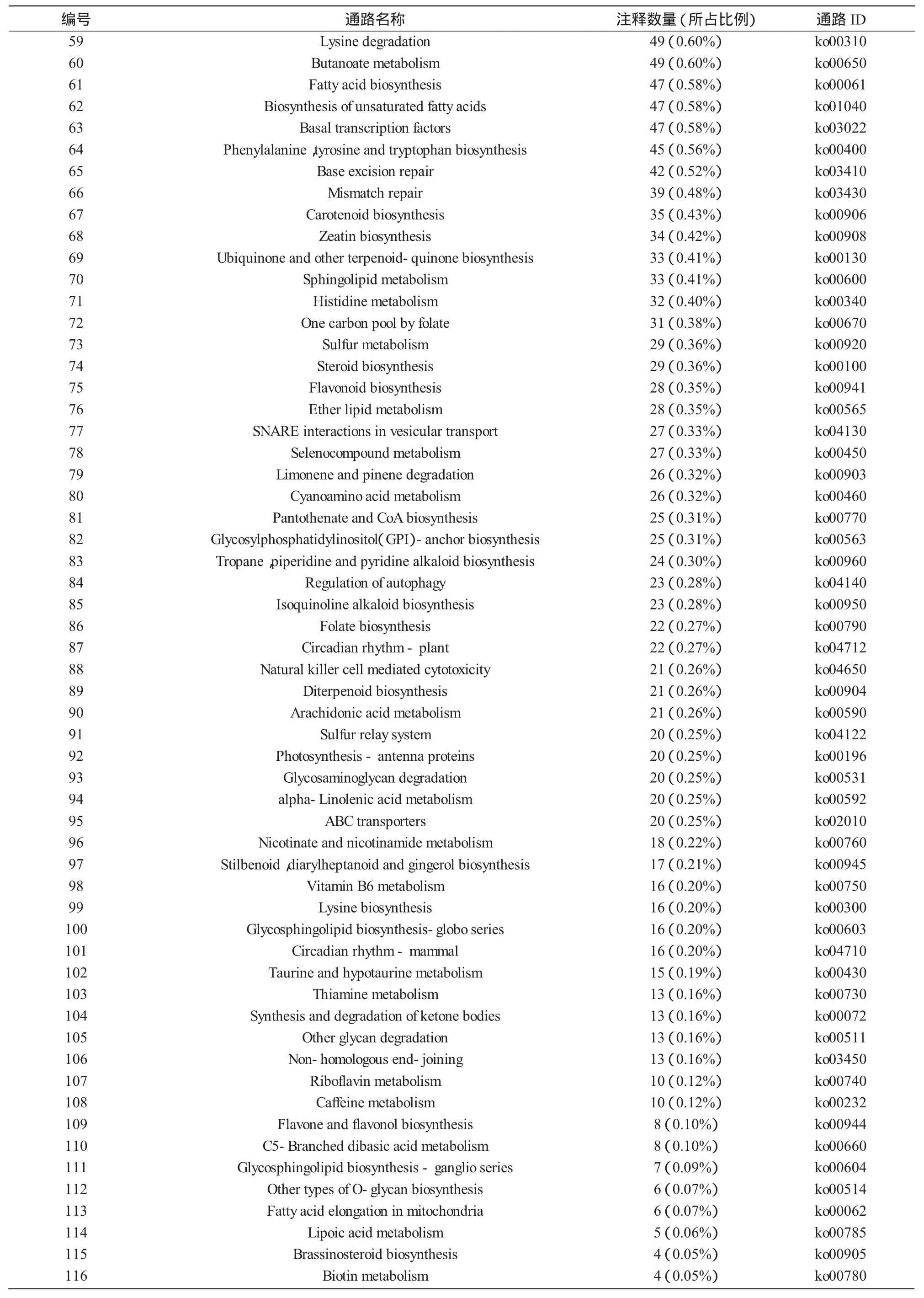
續表4
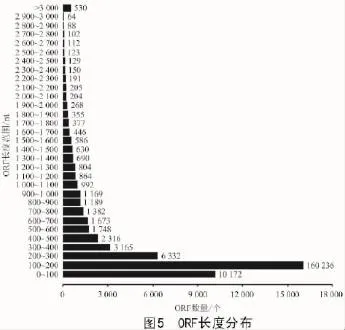

表5 SSR分析統計結果
3 討論
目前,RNA-seq技術已被廣泛應用于各物種的轉錄組分析中[15,11,17,20-21]。本試驗采用高通量測序技術對黃芩轉錄組進行測序,經過數據組裝共得到53 353條Unigenes,平均序列長度為797.64 bp,其中,長度在1 kb以上的Unigenes有13 439條。經過生物信息學分析,成功獲得功能注釋的Unigene有29 382條(55.07%),但仍有23 971條(44.93%)Unigene未能獲得注釋。分析其原因可能有2個:一是本試驗得到的Unigene序列長度較短,超過1/2的長度分布在500 nt以下,難以獲得同源性比對,這在一定程度上增加了基因功能注釋的難度;二是基因數據庫中的生物信息暫時缺乏,一些表達不豐富的基因可能無法獲得準確的功能注釋。本試驗所獲得的黃芩轉錄組數據,可豐富黃芩的基因組信息和公共數據庫信息,為進一步挖掘和鑒定出更多的黃芩功能基因奠定基礎。
本研究利用生物信息學手段對所有獲得的53 353條Unigenes進行了功能分類和代謝途徑分析。有10 756條Unigenes得到COG功能注釋,并歸入25種COG分類中,但仍有453條Unigenes未得到功能注釋,這可能與黃芩的基因組信息不完整有關。隨著更多物種基因組和轉錄組測序結果的出現,使公共數據庫不斷得到補充和完善,這些功能未知的基因將被準確注釋,并可能從中發現一些新的功能基因。另外,通過KEGG數據庫分析,發現了一些與黃芩有效成分合成有關的關鍵酶序列,這不僅為基因的克隆和功能研究提供了一定的基礎數據,還為進一步研究該有效成分生物合成途徑中關鍵基因的調控機制奠定基礎,同時也為應用生物學技術來合成黃芩中的有效成分提供可行性。
高通量測序技術與傳統測序方法相比,操作簡單,效率高,能夠挖掘出大量的SSR資源。本研究利用MISA軟件查找黃芩轉錄組測序數據,共發現了5 658個SSR位點,而且發現黃芩轉錄組SSR重復堿基以雙堿基型重復最多,占所有SSR的51.75%,這與其他研究結果類似[28-29]。黃芩SSR位點的發現可為黃芩分子標記的開發、群體遺傳多樣性分析、遺傳連鎖圖譜構建等后續試驗研究奠定理論基礎。本試驗對黃芩轉錄組進行了初步的探究,彌補了黃芩基因組信息十分缺乏的局面,為將來進行黃芩分子生物學方面的相關研究打下基礎,但要想深入了解黃芩的更多信息,今后仍需要加快黃芩轉錄組學和基因組學的相關研究步伐。
[1]李錫文,Hedge I C.中國植物志[M].北京:科學出版社&圣路易斯:密蘇里植物園出版社,1994.
[2]黃爽.神農本草經[M].北京:中醫古籍出版社,1982.
[3]中華人民共和國藥典委員會.中華人民共和國藥典 [M].北京:化學工業出版社,2005.
[4]中華本草編委.中華本草[M].上海:上海科學技術出版社,1998.
[5]Huang J M,Wang C J,Chou F P,et al.Protective effect of baicalin on tert-butyl hydroperoxide-induced rat hepatotoxicity[J].Arch Toxicol,2005,79(2):102-109.
[6]Zhang D Y,Wu J,Ye F,et al.Inhibition of cancer cell proliferation and prostaglandin E2 synthesis byScutellaria baicalensis[J].Cancer Res,2003,63(14):4037-4043.
[7]Marioni J C,Mason C E,Mane S M,et al.RNA-seq:An assessment oftechnical reproducibilityand comparison with gene expression arrays[J].Genome Res,2008,18(9):1509-1517.
[8]Fullwood M J,Wei C L,Liu E T,et al.Next-generation DNA sequencing of paired-end tags(PET)for transcriptome and genome analyses[J].Genome Res,2009,19(4):521-532.
[9]井趙斌,魏琳,俞靚,等.轉錄組測序及其在牧草基因資源發掘中的應用前景[J].草業科學,2011,28(7):1364-1369.
[10] Jewett M C,Oliveira A P,Patil K R,et al.The role of high-throughput transcriptome analysis in metabolic engineering [J].Biotechnol Bioproc Eng,2005,10:385-399.
[11]Donson J,Fang Y,Espiritu-Santo G,et al.Comprehensive gene expression analysis by transcript profiling[J].Plant Mol Biol,2002,48(1/2):75-97.
[12]Sultan M,Schulz MH,Richard H,et al.A global viewof gene activity and alternative splicing by deep sequencing of the human transcriptome[J].Science,2008,321:956-960.
[13]Wang E T,Sandberg R,Luo S,et al.Alternative isoform regulation in human tissue transcriptomes[J].Nature,2008,456:470-476.
[14]Birzele F,Schaub J,Rust W,et al.Into the unknown:Expression profiling without genome sequence information in CHO by next generation sequencing[J].Nucleic Acids Res,2010,38(12):3999-4010.
[15]Pan Q,Shai O,Lee L J,et al.Deep surveyingof alternative splicing complexity in the human transcriptome by high throughput sequencing[J].Nat Genet,2008,40(12):1413-1415.
[16]Nagalakshmi U,Wang Z,Waern K,et al.The transcriptional landscape ofthe yeast genome defined byRNA sequencing[J].Science,2008,320:1344-1349.
[17]Kakumanu A,AmbavaramMM,Klumas C,et al.Effects of drought on gene expression in maize reproductive and leaf meristem tissue revealed byRNA-Seq[J].Plant Physiol,2012,160(2):846-867.
[18]Filichkin S A,Priest H D,Givan S A,et al.Genome-wide mapping of alternative splicing in Arabidopsis thaliana[J].Genome Res,2010,20(1):45-58.
[19]WangL,Cao C,Ma Q,et al.RNA-Seq analyses of multiple meristems of soybean:novel and alternative transcripts,evolutionary and functional implications[J].BMCPlant Biol,2014,14:169.
[20]Vera J C,Wheat C W,Fescemyer H W,et al.Rapid transcriptome characterization for a nonmodel organismusing454 pyrosequencing [J].Mol Ecol,2008,17(7):1636-1647.
[21]郝大程,馬培,穆軍,等.中藥植物虎杖根的高通量轉錄組測序及轉錄組特性分析 [J].中國科學:生命科學,2012,42(5):398-412.
[22]李鐵柱,杜紅巖,劉慧敏,等.杜仲果實和葉片轉錄組數據組裝及基因功能注釋 [J].中南林業科技大學學報,2012,32(11):122-130.
[23]李瀅,孫超,羅紅梅,等.基于高通量測序454 GS FLX的丹參轉錄組學研究[J].藥學學報,2010,45(4):524-529.
[24]郭溆.基于轉錄組測序的石斛生物堿和人參皂苷生物合成相關基因的發掘、克隆及鑒定[D].北京:北京協和醫學院,2013.
[25]Grabherr M G,Haas B J,Yassour M,et al.Full-length transcriptome assembly from RNA-Seq data without a reference genome[J]. Nat Biotechnol,2011,29(7):644-652.
[26]G?tz S,García-Gómez J M,Terol J,et al.High-throughput functional annotation and data miningwith the Blast2GOsuite[J].Nucleic Acids Res,2008,36(10):3420-3435.
[27] Kanehisa M,Goto S.KEGG:Kyoto encyclopedia of genes and genomes[J].Nucleic Acids Res,2000,28:27-30.
[28]鄭紀偉.柳樹轉錄組高通量測序及SSR標記開發研究 [D].南京:南京林業大學,2013.
[29]陳浩東.達爾文氏棉旱脅迫轉錄組測序、EST-SSR開發及高密度遺傳圖譜構建[D].北京:中國農業科學院,2013.
Transcriptome Data Assembly and Analysis ofScutellaria baicalensisthrough High-throughput Sequencing
WANGDefu,CHENGYuanyuan,YANGFeng,NIUErbo,NIUYanbing
(College ofLife Sciences,Shanxi Agricultural University,Taigu 030801,China)
To study the transcriptome data of Scutellaria baicalensis Georgi,the root,stem,flower and leaf organs were used as the experimental material for the transcriptome sequencing,and analyzed by bioinformatics method.The transcriptome library of Scutellaria baicalensis contained 29 099 899 reads,and 53 353 Unigenes were obtained by assembling the Scaffolds in the transcriptome library; Unigene in the transcriptome of Scutellaria baicalensis could be divided into 25 classes according to the function by comparing Unigene and the COG database.The Unigene GO functions in the transcriptome library were classificated into 3 categories and 57 branches; Unigene in the transcriptome could be divided into116 classes accordingtothe metabolic pathway.Meanwhile,the paper alsogot number of 20 552 ORF using the GetORF software and got a number of 5 658 SSR markers by SSR analysis.Data presented in this study will constitute an important genomic resource for Scutellaria baicalensis Georgi and laya solid foundation for future gene clone and regulation research about the biosynthesis ofbaicalin.
Scutellaria baicalensis Georgi;transcriptome sequencing;bioinformatics analysis
S567.23+9
A
1002-2481(2016)08-1065-08
10.3969/j.issn.1002-2481.2016.08.04
2016-07-22
國家自然科學基金項目(31540050);山西農業大學科技創新基金項目(20142-10);山西農業大學引進人才科研啟動基金項目(2014YJ05)
王德富(1983-),男,甘肅慶陽人,講師,博士,主要從事分子植物病毒學研究工作。牛顏冰為通信作者。
猜你喜歡
中華詩詞(2022年6期)2022-12-31 06:41:24
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
電子制作(2018年18期)2018-11-14 01:48:24
中國科技論壇(2017年7期)2017-07-25 08:49:53
財經(2017年2期)2017-03-10 14:35:35
山東工業技術(2016年15期)2016-12-01 05:31:22
財經(2016年15期)2016-06-03 07:38:02
財經(2016年3期)2016-03-07 07:44:46
財經(2016年6期)2016-02-24 07:41:51
中國中醫藥現代遠程教育(2014年22期)2014-03-01 04:32:55
