分子印跡電化學傳感器的制備及其快速檢測飲水中草甘膦殘留的應用研究
2017-01-06 04:29:20李騰飛趙風年王珊珊佘永新金茂俊劉海金鄭鷺飛
分析測試學報 2016年12期
關鍵詞:檢測
張 超,李騰飛,趙風年,王珊珊,佘永新*,金茂俊,劉海金,金 芬,邵 華,鄭鷺飛,徐 平,王 靜*
(1.中國農業科學院 農業質量標準與檢測技術研究所,北京 100081;2.西藏自治區農畜產品質量安全檢驗檢測中心,西藏 拉薩 850000)
?
分子印跡電化學傳感器的制備及其快速檢測飲水中草甘膦殘留的應用研究
張 超1,李騰飛1,趙風年1,王珊珊1,佘永新1*,金茂俊1,劉海金2,金 芬1,邵 華1,鄭鷺飛1,徐 平2,王 靜1*
(1.中國農業科學院 農業質量標準與檢測技術研究所,北京 100081;2.西藏自治區農畜產品質量安全檢驗檢測中心,西藏 拉薩 850000)
以吡咯(Py)為功能單體,草甘膦(Gly)為模板,采用電化學聚合法構建了草甘膦分子印跡電化學傳感器。通過循環伏安法(CV)、差分脈沖伏安法(DPV)、電化學交流阻抗法(EIS)對印跡電極性能進行了表征,篩選了印跡電極的聚合體系和模板分子的洗脫方法,優化了檢測體系的pH值和吸附時間等。結果表明,以鐵氰化鉀為電活性探針,在最優檢測體系中該印跡傳感器對草甘膦具有特異性快速響應、靈敏度高和穩定性好的優點,傳感器的峰電流與草甘膦濃度在5~800 ng/mL范圍內呈良好的線性關系,相關系數(r2)為 0.981 7,檢出限(S/N=3)為0.27 ng/mL。該傳感器具有良好的重現性和穩定性,放置3周后對目標物的響應峰電流無明顯變化。用于實際樣品中草甘膦的測定,加標回收率為 78.6%~99.0%,能滿足現場快速檢測的要求。
吡咯;草甘膦;分子印跡電化學傳感器;飲水
草甘膦(Glyphosate,Gly)是一種內吸傳導型廣譜滅生性除草劑,其作用機理主要是抑制植物體內的烯醇丙酮基莽草素磷酸合成酶,從而抑制莽草素向苯丙氨酸、酪氨酸及色氨酸的轉化,使蛋白質合成受到干擾,導致植物死亡[1]。該類除草劑具有廣譜高效、低毒、易分解等特性,廣泛應用于果園、桑園、茶園、膠園等多種領域。目前,草甘膦是世界上應用最廣、產量最大的農藥品種,年銷售量高居農藥之首。2015年“草甘膦致癌風波”和2016年3月德國啤酒中被檢出草甘膦(含量在0.49~29.74 μg/L之間)等事件[2],進一步助推了國際社會對草甘膦安全風險的極大關注。中國是草甘膦的第一大生產國和出口國,隨著抗草甘膦植物的推廣和抗草甘膦雜草的增加,其使用量逐年增加,對農作物、生態環境和人類健康可能帶來一定的安全風險。因此,開展水環境中草甘膦殘留的研究具有重要現實意義。
世界各國對飲水中草甘膦的最大殘留量(MRL)均制定了限量規定,美國環境保護協會(US-EPA)制定飲水中草甘膦的MRL為700 μg/L[3];加拿大規定飲水中草甘膦最大可接受限量(MAC)為280 μg/L[4];德國規定飲用水中Gly極值不可超過0.1 μg/L[5];我國在《生活飲用水衛生標準》(GB5749-2006)規定了飲用水中Gly限量為700 μg/L[6]。目前,草甘膦的檢測方法主要有高效液相色譜法[7]、高效液相色譜-串聯質譜法[8]、氣相色譜法、氣相色譜-串聯質譜法[9]、毛細管電泳法[10]、離子色譜法[11]、酶聯免疫法[12]和電化學發光法[13]等。由于草甘膦極性較大,難溶于有機溶劑,缺乏可用于檢測的官能團,采用LC和GC色譜方法檢測均需衍生化;離子色譜法干擾因素較多,對草甘膦的檢測結果會產生一定的影響;ELASA等快速檢測技術存在穩定性差和靈敏度不高等缺點。因此,為滿足飲水中草甘膦的痕量檢測,研發操作簡便、成本低、靈敏度高、特異性好的快速檢測方法顯得尤為重要。
分子印跡聚合物(Molecularly imprinted polymers,MIPs)具有形狀、尺寸及功能基團與目標分子相匹配的印跡位點,對目標分子具有特異識別性能,因抗惡劣環境能力強、重復使用、穩定性好等優點,在傳感器技術、環境檢測、醫藥領域等方面展現出良好的應用前景[14-15]。MIPs作為特異性識別元件與電極相結合,可制備出對目標分子具有高度選擇識別性的分子印跡電化學傳感器,已被用于敵草隆[16]、環嗪酮[17]、綠麥隆[18]、速滅威[19]等多種農藥殘留的檢測。分子印跡傳感器通常存在著模板分子洗脫困難和聚合膜穩定性差的問題。吡咯具有良好的導電性、氧化還原性及局部交聯的特性,作為功能單體可以通過摻雜和脫摻雜形成穩定性好、吸附性能強的疏松多孔結構的聚吡咯分子印跡聚合膜,該聚吡咯分子印跡聚合膜具有模板分子快速固載、洗脫和傳質等優點。本文以吡咯為功能單體,草甘膦為模板分子,通過電化學聚合和聚吡咯過氧化法,在電極表面合成了聚吡咯多孔印跡膜,制備了草甘膦分子印跡電化學傳感器,以鐵氰化鉀為活性探針,篩選和優化關鍵參數,構建了草甘膦殘留檢測方法學體系,實現了飲水中草甘膦殘留的快速檢測。
1 實驗部分
1.1 儀器與試劑
CHI630E電化學工作站(上海辰華儀器公司);KQ2200DB3L超聲波清洗儀(昆山市超聲儀器有限公司);DF-101水浴鍋(鞏義市予華儀器有限公司);S-4800掃描隧道顯微鏡(日本Hitachi公司);Milli-Q超純水系統(法國Millipore公司)。采用三電極體系,金電極(直徑為3 mm)為工作電極,鉑網電極為對電極,飽和甘汞電極為參比電極(天津艾達恒晟科技有限公司)。
草甘膦、氨甲基膦酸、吡咯(阿拉丁試劑有限公司);毒死蜱、涕滅威(Dr.Ehrenstoefer GmbH);5 mmol/L鐵氰化鉀溶液:K3[Fe(CN)6]0.164 5 g,K4[Fe(CN)6] 0.211 2 g,KCl 0.745 5 g 溶于100 mL 0.01 mol/L PBS溶液;Britton-Robison緩沖溶液(BR):0.04 mol/L磷酸、硼酸和醋酸配成,用0.2 mol/L的NaOH調節pH值。所有實驗均在室溫下進行。
1.2 分子印跡電極及非印跡電極的制備
將金電極依次用 0.3,0.05 μm Al2O3粉拋光,再依次用無水乙醇和二次蒸餾水超聲清洗,每次5 min,用N2吹干。
采用循環伏安法在金電極表面制備分子印跡膜:聚合溶液含有13 μL吡咯(36.98 mol/L)、1 mL草甘膦(7.396 mol/L)和4 mL BR緩沖液(0.04 mol/L,pH 5.0),超聲2 min,在-1.0~1.0 V 電位范圍內,以100 mV·s-1速率循環掃描5圈。采用過氧化法去除模板分子:將所制電極置于0.1 mol/L NaOH溶液中,采用循環伏安法,在-1.3~1.2 V電位范圍內,以100 mV·s-1速率循環掃描 20 圈,形成過氧化聚吡咯,從而洗脫模板分子。采用相同的制備條件,不加入草甘膦模板,制備非印跡膜電極。制備過程如圖1所示。

圖1 草甘膦分子印跡電極的制備及檢測過程圖Fig.1 Schematic representation of the preparation of imprinted electrode and the process of detection
1.3 實驗方法
采用標準三電極檢測體系:印跡傳感器為工作電極,鉑絲電極為輔助電極,飽和甘汞電極為參比電極。循環伏安和差分脈沖伏安法(DPV)表征電極均采用含有 5 mmol/L K3Fe(CN)6的 0.5 mol/L KCl溶液。循環伏安法表征參數:起始電壓-0.2 V;終止電壓+0.6 V;掃速50 mV·s-1;差示脈沖法表征參數:起始電壓-0.2 V;終止電壓+0.6 V;掃速50 mV·s-1。
將去除模板分子的印跡電極先在空白底液0.2% 甲酸水溶液(pH 4.0)中進行多次CV掃描(-0.2~0.6 V),直至曲線穩定。然后將電極浸入含有一定濃度Gly的1/1 000甲酸水溶液(pH 4.0)中,富集18 min后,取出電極,用超純水反復沖洗電極表面,去除非特異性吸附到電極表面的物質,再將電極置入鐵氰化鉀溶液中進行電化學檢測。
2 結果與討論
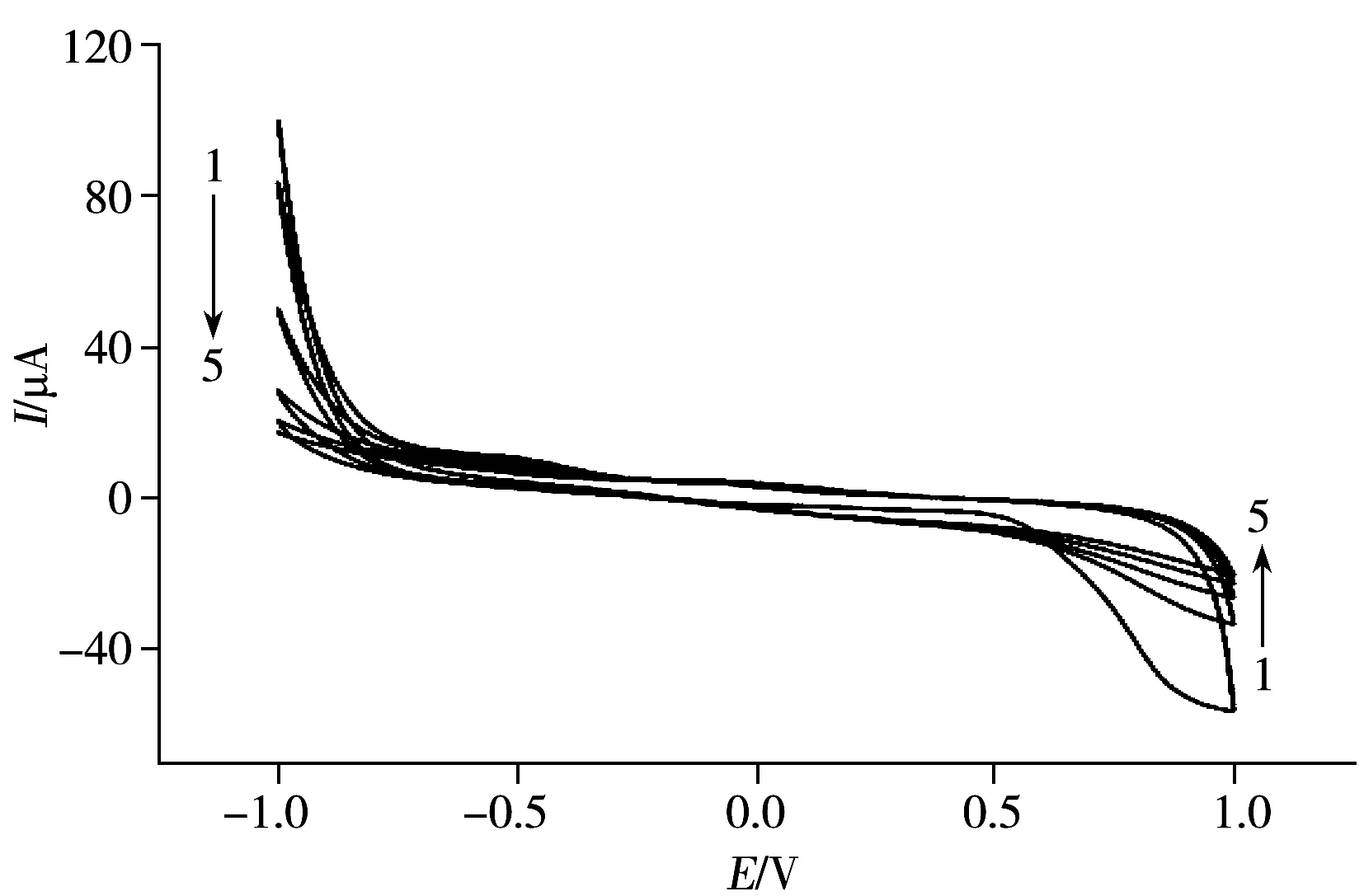
圖2 草甘膦分子印跡膜電聚合過程的循環伏安曲線Fig.2 Cyclic voltammograms for electropolymerization of imprinted film in the presence of glyphosate
2.1 分子印跡電極制備體系的優化
2.1.1 聚合體系篩選 以吡咯作為功能單體時,聚合物體系是影響陰離子的目標物摻雜過程的重要因素。本實驗研究了電極在BR緩沖液(pH 5.0)、0.2 mol/L硫酸和PBS緩沖液(pH 7.0) 3種聚合體系中的印跡效果。結果表明:工作電極在BR緩沖液(pH 5.0)聚合體系下制備的分子印跡膜對草甘膦具有很好的識別能力,此時草甘膦以負離子形式被成功摻雜到含正電聚吡咯網絡中,而在其他緩沖液條件下合成的膜對草甘膦無響應。因此,本實驗確定以BR緩沖液(pH 5.0)為最佳聚合體系。
2.1.2 聚合溶液模板與吡咯單體配比的選擇 功能單體與模板分子的比例對印跡膜的識別性能和形貌有影響。通過改變草甘膦與吡咯的濃度比(1∶3,1∶5,1∶8)制備了不同的印跡聚合膜,比較了這些電極在相同條件下分別吸附同一濃度(100 ng/mL)草甘膦溶液的效果。結果顯示,當功能單體與模板分子的比例為1∶5時,印跡電極對目標物的響應電流變化最大,此時印跡電極能夠形成更多有效的識別位點。

圖3 草甘膦分子印跡膜洗脫過程的循環伏安曲線Fig.3 Cyclic voltammograms for the elution of imprinted electrode

圖4 不同電極在含 5 mmol/L K3Fe(CN)6的0.5 mol/L KCl溶液中的循環伏安圖Fig.4 Cyclic voltammograms of different electrodes in solution containing 5 mmol/L K3Fe(CN)6and 0.5 mol/L KCla.bare gold electrode;b.MIP-modified gold electrode after elution;c.MIP-modified gold electrode/NIP-modified gold electrode

圖5 不同草甘膦分子印跡電極的交流阻抗圖Fig.5 Electrochemical impedance spectra of different electrodesa.bare gold elecrode,b.MIP-modified gold electrode after elution,c.NIP-modified gold electrode,d.MIP-modified gold electrode;insert:partial enlarged drawing of a and b
2.1.3 印跡電極的循環伏安特性及電化學行為表征 圖2是含模板分子Gly的聚合液在金電極表面聚合過程中的CV曲線。由圖可知,從第2圈開始,在電位為-0.3 V和+0.9 V處,金電極在聚合體系中的氧化還原峰電流均逐漸減小,隨著掃描圈數的增加,金電極的氧化還原峰電流緩慢減小,直至曲線趨于平直,說明在電位-1.0~+1.0 V之間,聚吡咯膜由于陰離子模板分子的摻雜,在金電極表面逐漸由導電膜轉變為鈍化的不導電聚合膜,阻礙了溶液與電極之間的電子傳遞,從而使電流響應值降低。
當聚吡咯印跡電極在-1.3~1.2 V電位范圍內,浸入 0.1 mol/L NaOH溶液中進行循環伏安掃描而被過氧化后,由于溶液中的強親核試劑OH-與聚吡咯環上活性位點的相互作用,促使含氧基團羰基或羧基被引入聚吡咯結構單元,對陰離子模板分子產生排斥,實現了模板分子的脫摻雜[20]。圖3是印跡電極洗脫過程的CV曲線。由圖可見0.8~1.0 V 是PPy的氧化電位,隨著掃描圈數的增加,-0.3 V與0.9 V 處的電流逐漸增大,說明聚吡咯膜在堿性條件下被過氧化后可促進Gly模板分子脫摻雜過程,形成了新的電子傳遞通道[21]。
以 5 mmol/L K3Fe(CN)6(含0.5 mol/L KCl)溶液為電化學信號探針,采用循環伏安法對不同電極進行表征,如圖4所示。裸金電極(曲線a)有最大的氧化還原峰電流,印跡電極聚合曲線和非印跡電極聚合曲線(曲線c)重合,幾乎無峰電流。而洗脫模板分子后的印跡電極(曲線b)峰電流介于曲線a與曲線c之間,說明洗脫模板分子后,形成的印跡空穴形成了電子傳遞通道,有利于探針在印跡電極表面發生氧化還原反應而產生信號。
本實驗進一步采用電化學阻抗技術(EIS)對不同電極進行表征。在 EIS 圖中,半圓的直徑越大,表示其阻抗越大,相應的電子轉移越難。如圖5所示,裸金電極(曲線a)有很大的峰電流絕對值,其EIS半徑最小,交流阻抗最小,這是因為裸金電極表面無阻止探針電子傳遞的聚合膜;非印跡電極(曲線c)與印跡電極(曲線d)由于表面均覆蓋了鈍化的聚合膜,電子探針轉移受阻,所以EIS半徑均比裸金電極大。而印跡電極(曲線d)的EIS半徑遠大于非印跡電極(曲線c)的EIS半徑,這是由于草甘膦是非電活性物質,被摻雜于聚吡咯骨架中阻礙了電子的傳遞,其阻抗增加。而洗脫模板分子后電極留下的“孔穴”使部分電子探針到達電極表面,所以洗脫后印跡電極(曲線b)的EIS半徑小于非印跡電極的半徑,表明其電子轉移阻抗比非印跡電極阻抗小。
2.2 實驗條件的優化
2.2.1 檢測體系及pH值的影響 分別配制含1 μg/mL草甘膦的各種溶液(BR緩沖液、PBS緩沖液和甲酸水溶液),各溶液pH值為2.0,3.0,4.0,5.0,6.0,7.0,將印跡電極在相同條件下進行吸附檢測,結果表明印跡電極在含草甘膦的甲酸水溶液中檢測信號最高,在其他溶液中對草甘膦基本無響應。因此,選擇甲酸水溶液作為檢測草甘膦的溶液體系。由于不同的pH值條件下,溶液中草甘膦所帶的電荷不同,電極對草甘膦的響應也不同。因此本實驗研究了印跡電極在不同pH值(3.0,4.0,5.0,6.0,7.0)甲酸水溶液中對草甘膦的響應。結果表明,當溶液的pH值為4.0時,印跡電極對草甘膦的吸附最大,其探針分子的電流值最小;隨著體系pH值增大,印跡電極對草甘膦的吸附減小,其探針分子的響應電流值增大,說明pH值對印跡電極檢測草甘膦具有重要的影響,推測可能是隨著pH值增加,溶液中的草甘膦分子帶負電荷增多,與過氧化聚吡咯膜帶負電的羰基和羧基產生靜電排斥,從而導致吸附減少,探針分子電流增大[22]。因此,實驗選擇pH 4.0作為檢測草甘膦的最佳酸堿度。
2.2.2 吸附時間的影響 采用差分脈沖伏安法(DPV)考察了50 ng/mL草甘膦在洗脫模板后的分子印跡電極上峰電流隨時間的變化情況。結果顯示,隨著吸附時間的延長,印跡電極對草甘膦的吸附量增加,其探針分子的峰電流逐漸降低,當18 min之后峰電流幾乎不變。因此,選擇18 min作為檢測體系的最佳吸附時間。

圖6 印跡電極的吸附和洗脫效果Fig.6 Absorption and elution of imprinted electrode towards glyphosatea:NIP-modified gold electrode after elution;b:MIP-modified gold electrode after elution;c:NIP-modified gold electrode after absorption;d:MIP-modified gold electrode after absorption
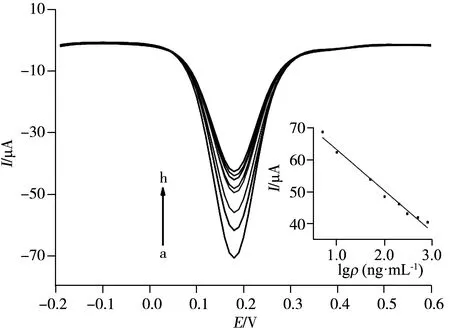
圖7 吸附不同濃度草甘膦的分子印跡傳感器在鐵氰化鉀溶液中的DPV圖Fig.7 DPV curves of MIP-modified gold electrode in different concentration of Gly concentration of Gly(a-h):5,10,50,100,200,300,500,800 ng/mL;5.0 mmol/L K3[Fe(CN)6],0.1 mol/L KCl;insert:calibration curve of glyphosate
2.3 印跡電極與非印跡電極吸附的對比
將印跡電極與非印跡電極在相同條件下吸附50 ng/mL草甘膦18 min后,觀察吸附和洗脫模板分子前后DPV的變化(圖6)。結果顯示,非印跡電極對草甘膦的響應微小,而印跡電極由于吸附了草甘膦其探針分子的電流發生了很大變化,當模板分子洗脫后,印跡電極的探針響應電流又恢復,說明印跡電極對草甘膦具有特異性吸附性能,也表明了洗脫條件的效果。
2.4 方法的線性范圍與檢出限
采用差分脈沖伏安法(DPV)考察了分子印跡電極吸附不同濃度的草甘膦18 min后的峰電流變化趨勢(圖7),草甘膦濃度(ρ,ng/mL)在5~800 ng/mL范圍內與峰電流(I)呈良好的線性關系,線性方程為I=-12.86lgρ+75.92,r2=0.981 7,檢出限(S/N=3)為0.27 ng/mL。
2.5 電極的選擇性
為研究修飾電極對草甘膦的選擇性,采用差分脈沖伏安法測定印跡傳感器和非印跡傳感器對1 000 ng/mL草甘膦、氨甲基膦酸(AMPA)、毒死蜱(Chlorpyrifos)、涕滅威(Aldicarb)的吸附能力。實驗結果表明,印跡電極對草甘膦的吸附最大,對毒死蜱和氨甲基膦酸有少量吸附,這可能是印跡識別位點與磷酸根有關,而對于結構差異較大的涕滅威幾乎不吸附;非印跡電極對以上農藥幾乎無吸附,這說明印跡傳感器對于草甘膦有很好的特異性識別性能,盡管對以上結構類似物有少量吸附,但不影響傳感器對草甘膦的檢測。
2.6 電極的重現性與穩定性
在相同條件下制備5支印跡電極,對50 ng/mL 草甘膦溶液平行測定5次,計算電流響應值的相對標準偏差為3.1%,表明該印跡傳感器具有良好的重現性,分子印跡電極經洗脫可重復利用,表明模板分子與印跡膜上“孔穴”的結合為可逆過程,印跡電極可重復使用。電極經多次使用,洗脫后置于超純水中常溫保存,3周后其響應值降至初始響應值的80.9%,表明印跡電極的穩定性良好。
2.7 實際樣品的加標檢測
為了評價本方法的實用性,隨機選擇9份自來水進行加標回收率實驗。分別取100,200,500 μL的1 μg/mL草甘膦標準溶液用自來水定容至10 mL,加標濃度分別為10,20,50 ng/mL。不做任何前處理,直接用分子印跡電極檢測18 min,每個樣品平行測定3次。結果測得草甘膦在自來水樣品中的回收率為78.6%~99.0%,相對標準偏差為2.8%~4.5%,表明草甘膦分子印跡電化學傳感器可以實現自來水中草甘膦農藥殘留的快速檢測。
3 結 論
本文以吡咯為功能單體,草甘膦為模板分子,通過電化學聚合和聚吡咯過氧化法,在電極表面合成了聚吡咯多孔印跡膜,制備了草甘膦分子印跡電化學傳感器。該傳感器制備方便,模板分子摻雜和洗脫簡單,對目標物的特異性強、穩定性好、傳質速度快、響應時間短(僅為18 min)。在5~800 ng/mL草甘膦濃度范圍內,傳感器響應值與草甘膦濃度的對數值呈良好的線性關系,其檢出限為0.27 ng/mL,對結構類似物等干擾物質具有很強的抗干擾能力,可滿足飲水中痕量草甘膦的檢測要求。
[1] Duke S O,Powles S B.PestManag.Sci.,2008,64(4):319-325.
[2] Guyton K Z,Loomis D,Grosse Y,El Ghissassi F,Benbrahim-Tallaa L,Guha N,Scoccianti C,Mattock H,Straif K.LancetOncol.,2015,16(5):490-491.
[3] Horciciak M,Masar M,Bodor R,Danc L,Bel P.J.Sep.Sci.,2012,35(5/6):674-680.
[4] Coutinho C F B,Coutinho L F M,Mazo L H,Nixdorf S L,Camara C A P.J.Chromatogr.A,2008,1208(1/2):246-249.[5] Popp M,Hann S,Mentler A,Fuerhacker M,Stingeder G,Koellensperger G.Anal.Bioanal.Chem.,2008,391(2):695-699.
[6] GB 5749-2006.Standards for Drinking Water Quality.National Standards of the People's Republic of China(生活飲用水衛生標準.中華人民共和國國家標準).
[7] Fang F,Xu H,Wei R Q,Liu X N,Xu R,Li S C,Liu B K.J.Instrum.Anal.(方芳,徐會,魏榮卿,劉曉寧,徐蓉,李壽椿,劉寶菎.分析測試學報),2011,30(6):683-686.
[8] Zhou S,Xu D M,Lin L Y,Chen L P,Zhou Y,Yang L Z.J.Instrum.Anal.(周爽,徐敦明,林立毅,陳鷺平,周昱,楊黎忠.分析測試學報),2013,32(2):199-204.
[9] Steinborn A,Alder L,Michalski B,Zomer P,Bendig P,Martinez S A,Mol H G J,Class T J,Costa P N.J.Agric.FoodChem.,2016,64(6):1414-1421.
[10] Lanaro R,Costa J L,Cazenave S O S,Zanolli-Filho L A,Tavares M F M,Chasin A A M.J.ForensicSci.,2015,60(S1):241-247.
[11] Zhang P Z,Wu J,Zhang P M,Xu Y.J.Instrum.Anal.(張培志,吳軍,張培敏,徐育.分析測試學報),2003,22(4):89-90.
[12] Selvi A A,Sreenivasa M A,Manonmani H K.FoodAgric.Immunol.,2011,22(3):217-228.
[13] Cai Q,Chen X,Qiu B,Lin Z.Chin.J.Chem.,2011,29(3):581-586.
[14] Yao T,Gu X,Li T F,L J G,L J,Zhao Z,Wang J,Qin Y C,She Y X.Biosens.Bioelectron.,2016,75:96-100.[15] She Y X,Cao W Q,Shi X M,Lv X L,Liu J J,Wang R Y,Jin F,Wang J,Xiao H.J.Chromatogr.B,2010,878(23):2047-2053.
[16] Wong A,Sotomayor M D P T.J.Electroanal.Chem.,2014,731:163-171.
[17] Toro M J U,Marestoni L D,Sotomayor M D P T.Sens.ActuatorB,2015,(208):299-306.
[18] Li X,Zhang L M,Wu C R,Wei X P,Li J P.J.Instrum.Anal.(李雪,張連明,吳昌儒,魏小平,李建平.分析測試學報),2013,32(11):1344-1348.
[19] Pan M F,Fang G Z,Liu B,Qian K,Wang S.Anal.Chim.Acta,2011,690(2):175-181.
[20] Sahin M,Sahin Y,Ozcan A.Sens.ActuatorB,2008,133:5-14.
[21] Susana M,Otto S W.Anal.Chim.Acta,1996,334:149-153.
[22] McConnell J S,Hossner L R.J.Agric.FoodChem.,1985,6(33):1075-1078.
Preparation of an Electrochemical Sensor Based on Molecularly Imprinted Polymer and Its Application in Determination of Glyphosate Residues in Water Samples
ZHANG Chao1,LI Teng-fei1,ZHAO Feng-nian1,WANG Shan-shan1,SHE Yong-xin1*,JIN Mao-jun1,LIU Hai-jin2,JIN Fen1,SHAO Hua1,ZHENG Lu-fei1,XU Ping2,WANG Jing1*
(1.Institute of Quality Standards&Testing Technology for Agri-Products,Chinese Academy of Agricultural Sciences,Beijing 100081,China;2.Tibet Testing Center of Quality and Safety for Agricultural and Animal Husbandry Products,Lhasa 850000,China)
A novel ultra-sensitive molecularly imprinted electrochemical senor with good imprinted capacity to glyphosate was prepared by electropolymerisation on the gold electrode surface,with pyrrole(Py) as functional monomer,glyphosate(Gly) as template.The prepared electrode was characterized by cyclic voltammetry(CV),differential pulse voltammetry(DPV) and electrochemical impedance spectroscopy(EIS).The condition of polymerization,the method of eluting template,the pH value of detection andincubation time were optimized.The optimal conditions were as follows:supporting electrolyte:Britton Robison buffer solution(BR,pH 5.0),ratio of template to functional monomers:1∶5,incubation system:formic acid aqueous solution(pH 4.0),incubation time:18 min.Under the optimum experimental conditions,the prepared electrode showed rapid response,high sensitivity and good selectivity for the template molecule glyphosate.A good linear relationship between oxidation peak current and Gly concentration was obtained over the range of 5-800 ng/mL with a correlation coefficient of 0.981 7 and a detection limit(S/N=3) of 0.27 ng/mL.The prepared sensor also showed good reproducibility and stability,the sensor was successfully applied in the determination of glyphosate in the tap water with recoveries of 78.6%-99.0%.
pyrrole;glyphosate;molecularly imprinted electrochemical sensor;tap water
2016-05-19;
2016-06-30
國家自然科學基金項目(31471654);“十二五”國家科技支撐計劃項目( 2014BAD13B05-05)
10.3969/j.issn.1004-4957.2016.12.005
O657.1;S482.4
A
1004-4957(2016)12-1542-06
*通訊作者:佘永新,研究員,研究方向:仿生識別材料與檢測技術,Tel:010-82106513,E-mail:0891syx@163.com 王 靜,教授,研究方向:仿生識別材料與檢測技術,Tel:010-82106568,E-mail:wjing_2001@163.com
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
