氣相色譜-質譜法測定食品中黃樟素及其衍生物含量
2017-01-06 04:29:24祝偉霞楊冀州張淑霞
分析測試學報 2016年12期
關鍵詞:方法
祝偉霞,李 睢,張 麗,胡 鍇,楊冀州,張淑霞
(1.河南出入境檢驗檢疫局,河南 鄭州 450003;2.河南中醫藥大學 藥學院,河南 鄭州 450000)
?
氣相色譜-質譜法測定食品中黃樟素及其衍生物含量
祝偉霞1*,李 睢2,張 麗1,胡 鍇2,楊冀州1,張淑霞1
(1.河南出入境檢驗檢疫局,河南 鄭州 450003;2.河南中醫藥大學 藥學院,河南 鄭州 450000)
建立了調味制品(調味料、番茄醬、醬油、花椒)、飲料(碳酸飲料、乳飲料、果汁)、肉制品(香腸、肉罐頭、肉湯)、水果制品(果凍、果醬)、水產品(干魚肉、魚罐頭、魚肉泥)、小吃類(油炸薯片、糖果、巧克力)等18種食品中黃樟素、異黃樟素、二氫黃樟素含量的分析方法。樣品在氯化鈉鹽析作用下,經二氯甲烷-乙腈提取,無水硫酸鈉-氟羅里硅土柱吸附極性物質和水分。復雜樣品需再經PRiME HLB固相萃取柱(SPE)凈化,5%苯基-95%甲基聚硅氧烷毛細管氣相色譜柱分離,選擇離子質譜技術監測,內標法定量。在最佳實驗條件下,3種黃樟素在25~5 000 μg/kg范圍內呈良好的線性關系,相關系數(r2)≥0.998 1,方法檢出限(LOD)和定量下限(LOQ)分別為20 μg/kg和50 μg/kg,3個加標水平的回收率為77.6%~100.9%,相對標準偏差(RSD)為3.7%~13.6%。多種實際樣品的測定結果表明,該方法準確可靠,適用于食品中3種黃樟素及其衍生物的定量和定性分析。
氣相色譜-質譜(GC-MS);選擇離子掃描(SIM);固相萃取(SPE);黃樟素;香料添加劑;食品
黃樟素(SFL)廣泛存在于黃樟、沉水樟和堅葉樟等天然芳香植物提取物中,是黃樟精油、八角精油和樟腦油的主要成分,可作為天然香精用于食品、化妝品和煙草制品生產[1]。異黃樟素(DSFL)和二氫黃樟素(ISFL)是黃樟素的衍生物,也可作為香料添加劑用于食品加工[2]。研究表明,黃樟素對人體具有一定的毒副作用,易誘發基因突變和肝損傷,是消化系統、血液系統、泌尿系統的強致癌物[3-4]。因此,美國食品法規(§189.180)規定不得將黃樟素、異黃樟素、二氫黃樟素作為添加劑加入食品中[5],歐盟法規(EC/1334/2008)禁止將黃樟素作為香料和食品添加劑用于食品生產。為了控制天然植物或精油中污染黃樟素,歐盟還規定了天然食品中黃樟素的最高殘留水平,其中肉及肉制品為15 mg/kg,水產品及其制品為15 mg/kg,調味料及調味醬為25 mg/kg,不含酒精的飲料為1 mg/kg[6]。我國《化妝品衛生規范》(2015)禁止添加黃樟素。為保障我國食品安全,建立一種食品中黃樟素、異黃樟素和二氫黃樟素的分析方法是非常必要的。
目前,有關黃樟素檢測的研究主要涉及香精香料[7-9]、化妝品[10]、煙草制品[2,11]、天然產物[1,12],分別采用氣相色譜氫火焰離子化法(GC-FID)[13-14]、氣相色譜-質譜法(GC-MS)[9-11]、液相色譜熒光法(HPLC-FLD)[8]、液相色譜-質譜法(LC-MS)[7]等技術。2003年,Siano等[15]首次研究了GC-FID測定食品中黃樟素、甲基胡椒酚和甲基丁香酚的分析方法,目前尚無有關食品中黃樟素、二氫黃樟素、異黃樟素測定的報道。本方法以彌補食品中黃樟素及其衍生物檢測方法空白為目標,建立了固相萃取/氣相色譜-質譜技術測定18種食品中黃樟素及其衍生物的確證分析方法,可為食品質量安全和風險評估提供技術支持。
1 實驗部分
1.1 儀器與試劑
GC-MS QP2010plus氣相色譜-質譜儀(配電子轟擊源,日本Shimadzu公司);氟羅里硅土柱(6 mL/1 g,北京振翔工貿有限公司);OASIS PRiME HLB柱(6 mL/200 mg,美國Waters公司);5210型超聲波清洗器(美國Bransonic公司);SA-31振蕩器(日本大和科學株式會社);24位固相萃取裝置(德國CNW Technologies公司);CF15RXII離心機(日本Hitachi公司);N-EVAP112氮吹儀(美國Organomation Associates公司);0.2 μm有機相濾膜(天津津騰公司);二氫黃樟素(純度≥95%,德國Dr.Ehrenstorfer公司),黃樟素和異黃樟素純度均≥95%,購于加拿大TRC研究院;內標物質:3-間甲苯乙酸酯(純度99%,上海TCI化成工業公司);甲醇、二氯甲烷、乙腈(色譜純,美國Fisher公司);氯化鈉;無水硫酸鈉在650 ℃灼燒4 h,于干燥器中儲存備用。
1.2 溶液的配制
標準儲備溶液:分別稱取50 mg(精確至0.1 mg)標準物質,用甲醇溶解并定容至50 mL,配制成質量濃度為1.0 g/L的單個標準儲備液。該溶液于-18 ℃密封儲存,有效期6個月。內標儲備溶液:準確稱取10 mg(精確至0.1 mg)內標,用甲醇溶解并定容至100 mL,配制成質量濃度為100.0 mg/L的標準儲備液,該溶液于-18 ℃密封儲存,有效期6個月。混合標準溶液:分別量取1 mL標準儲備液于100 mL容量瓶中,用甲醇定容,配制成濃度為10.0 mg/L的混合標準溶液,于2~4 ℃密封儲存,有效期6個月。二氯甲烷-乙腈溶液(體積比1∶1):分別量取500 mL二氯甲烷和500 mL乙腈混合均勻后備用。
1.3 樣品前處理
1.3.1 飲料和水果制品 準確稱取2.5 g均勻試樣(精確至0.01 g)于50 mL聚丙烯離心管中,加入50 μL內標溶液(10.0 mg/L)、1.0 g氯化鈉和15 mL二氯甲烷-乙腈溶液,渦旋混勻3 min,超聲提取15 min,于4 000 r/min轉速下離心5 min,轉移上清液于另一離心管中。再加入15 mL二氯甲烷-乙腈溶液重復渦旋、超聲與離心步驟,合并兩次上清液,待凈化。
稱取1.0 g無水硫酸鈉裝載至Florisil柱中,轉移樣液至Florisil柱,待樣液完全通過后,加入5 mL乙腈淋洗,收集全部流出液于常溫下氮吹至20 mL左右,并用乙腈定容至25 mL。取1 mL樣液經0.22 μm濾膜過濾后,進行GC-MS測定。
1.3.2 調味制品、肉制品、水產品和小吃類食品 萃取步驟同上,合并兩次上清液,待凈化。
稱取1.0 g無水硫酸鈉裝載至Florisil柱中,轉移樣液至Florisil柱,待樣液流完全通過后,加入5 mL乙腈淋洗,收集全部流出液于常溫下氮吹至20 mL左右,并用乙腈定容至25 mL。吸取5 mL樣液過PRiME HLB 固相萃取柱,控制流速小于1.0 mL/min,棄去前段約0.5 mL流出液,收集1 mL流出液經0.22 μm濾膜過濾后,進行GC-MS測定。
1.4 GC-MS 條件
色譜柱:TR-5MS 5%苯基-95%甲基聚硅氧烷色譜柱,30 m×0.25 mm(i.d),0.25 μm;載氣:氦氣,純度≥99.999%;流速:恒流模式 1.0 mL/min;色譜柱升溫程序:初始溫度90 ℃保持1 min,以2.5 ℃/min升至 140 ℃,再以30 ℃/min升至250 ℃保持5 min。進樣口溫度:260 ℃;GC-MS接口溫度:250 ℃;進樣量:1 μL;進樣方式:不分流進樣;離子源:電子轟擊源(EI);電離能量:70 eV;離子源溫度:230 ℃;溶劑延遲時間:7.0 min;檢測方式:選擇離子監測(SIM);內標物監測離子:m/z108(定量離子),150,107,79;黃樟素和異樟素監測離子:m/z162(定量離子),131,104,77;二氫黃樟素監測離子:m/z164(定量離子),135,77,105。
2 結果與討論
2.1 提取溶劑的選擇
測定黃樟素、異黃樟素、二氫黃樟素的提取溶劑主要采用二氯甲烷和甲醇[7,10,13,15]。本方法對比了二氯甲烷和甲醇提取18種食品的效果。結果顯示,采用二氯甲烷提取含有乳蛋白基質的樣品時,易產生乳化現象,采用加熱、增加氯化鈉量、高速離心等方法均未能解決乳化問題;二氯甲烷提取巧克力、油脂等脂溶性樣品時,樣品全部溶解。進行GC-MS測定時,分析物的保留時間產生漂移,不利于定性和定量分析;而采用甲醇提取含水量高的食品時,需增加除水步驟,不利于氣相色譜測定。
實驗進一步考察了乙腈、乙酸乙酯、二氯甲烷、二氯甲烷-乙腈、正己烷作為提取溶劑時的提取效率。結果表明,以上溶劑對食品中黃樟素、異黃樟素、二氫黃樟素的提取效率均不低于90%,但采用乙酸乙酯、二氯甲烷、正己烷提取含乳蛋白或高含量淀粉的樣品時,易產生乳化現象,不利于實驗操作的進行。結合凈化方法的優化結果,本方法最終選用二氯甲烷-乙腈(1∶1)作為提取溶劑。
2.2 提取方式的選擇
以黃樟素陽性的調料粉、陰性樣品添加回收為實驗對象,考察了超聲、振蕩加熱、渦旋混合、超聲+渦旋振蕩等常用提取方法的效果。結果顯示,由于黃樟素類化合物具有揮發性,其在加熱條件下的回收率偏低;而采用超聲+渦旋振蕩時的回收率最高,因此本方法采用超聲+渦旋振蕩的方法進行提取。
2.3 凈化方法的選擇
本方法的18種基質可歸為高水分、高蛋白、高脂類、高糖類樣品,二氯甲烷-乙腈提取液未凈化直接測定時干擾物多。因此對凈化方法進行了研究,并考察了以下幾種凈化方法的回收率:液液萃取法(LLE)(飽和氯化鈉水溶液中二氯甲烷液液分配NaCl LLE、硅藻土液液萃取DmE LLE);正相SPE法(Florisil柱、硅膠柱Si、氨基柱NH2、Cleanert柱、中性氧化鋁柱Alumina-N);QuERChERS法;反相SPE法(HLB柱、PRiME HLB柱)。
實驗結果表明:LLE法僅限于測定液態和水溶性固態樣品,對于脂溶性固態樣品無法操作;采用硅膠固相萃取柱、氨基柱、Cleanert 柱、中性氧化鋁柱凈化食品中黃樟素、異黃樟素、二氫黃樟素時,3種分析物均未能定量保留和洗脫;QuERChERS法凈化后進行GC-MS測定,黃樟素和二氫黃樟素出峰處有干擾;樣液過Florisil柱過程中,極性干擾物被吸附,3種待測物全部流出,回收率最高,因此本方法采用Florisil柱去除干擾物。反相SPE法測定3種分析物的回收率均不小于53.2%,其中新型PRiME HLB柱的回收率最高。PRiME HLB柱可去除基質中蛋白、鹽、磷脂等干擾物,并以乙腈載體溶劑中效果最佳,因此對于復雜的食品樣品,本方法增加PRiME HLB進行凈化(圖1列出不同凈化方法對比結果)。
2.4 樣品濃縮方法的優化
比較了100 μg/L混合標準溶液未氮吹、氮吹過半、常溫氮吹至干、50 ℃氮吹至干的響應強度。結果發現內標物和3種分析物在未氮吹、氮吹過半中的響應強度未發生變化,氮吹至干后響應強度降低18.2%~31.5%,加熱氮吹至干后響應強度降低46.1%~63.0%,證明4種化合物在加熱或氮吹至干條件下易揮發,不利于準確定量。同時在采用PRiME HLB柱凈化過程中,溶液中二氯甲烷影響脂類物質的去除,因此本法采用常溫下氮吹去除樣液中二氯甲烷溶劑。
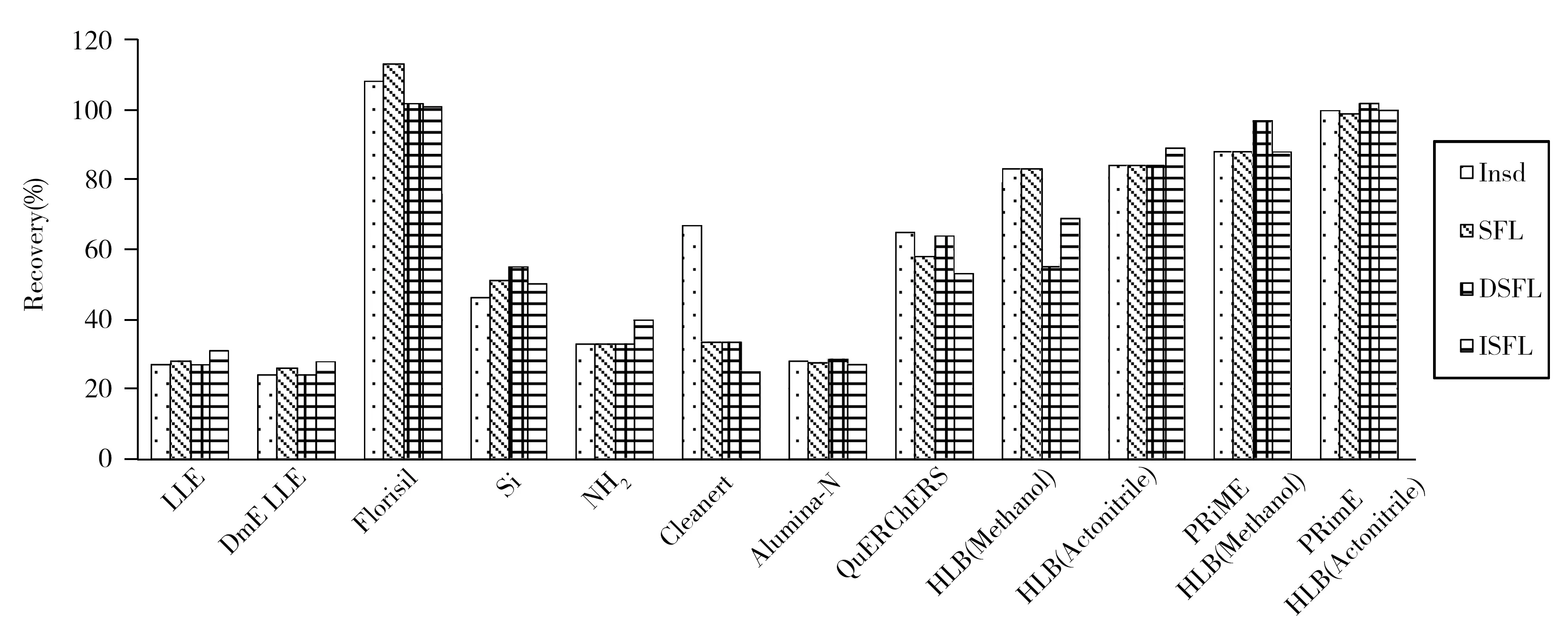
圖1 不同凈化方法的回收率比較Fig.1 Comparison on recoveries of different purification method
2.5 方法的線性關系、檢出限、回收率及精密度

圖2 標準溶液的總離子流色譜圖(50 μg/L)Fig.2 Total ion chromatogram of standard solution(50 μg/L)
采用乙腈配制2.5,5.0,10,25,50,100,500 μg/L的標準溶液,添加內標濃度為50 μg/L,以待測物與內標物的峰面積比為縱坐標(Y),待測物與內標的濃度比為橫坐標(X),繪制標準曲線,獲得線性方程:黃樟素Y=1.602×10-2X+1.671×10-3(r2=0.999 5),二氫黃樟素Y=8.694×10-3X+1.859×10-3(r2=0.998 1),異樟素Y=1.145×10-2X+1.893×10-3(r2=0.999 0)。由于采用本方法測定時,分析物被稀釋10倍,因此方法的線性范圍為25~5 000 μg/kg。標準溶液的SIM圖見圖2。將信噪比(S/N)≥3定義為LOD,S/N≥10定義為LOQ,得本方法的LOD為20 μg/kg,LOQ為50 μg/kg。
以不含黃樟素、異黃樟素、二氫黃樟素的調味制品(調味料、醬油、番茄醬、花椒)、飲料類(碳酸飲料、乳飲料、果汁)、肉制品(香腸、肉罐頭、肉湯)、水果制品(果凍、果醬)、水產品(干魚肉、肉泥、魚罐頭)、小吃類(薯片、糖果、巧克力)為實驗樣品,分別添加50,100,1 000 μg/kg 3個濃度水平進行回收實驗,每個濃度平行測定6次,測得平均回收率為77.6%~100.9%,相對標準偏差(RSD)為3.7%~13.6%。表1列出了部分調味制品中黃樟素、異黃樟素、二氫黃樟素的回收率和RSD。

表1 方法的加標回收率及相對標準偏差(n=6)
(續表1)

SampleAdded(μg/kg)SafroleIsosafroleDihydrosafroleRecovery(%)RSD(%)Recovery(%)RSD(%)Recovery(%)RSD(%)Chocolate508611078817686794100918104913419626810009021108458493363

圖3 方便面樣品中黃樟素(370 μg/kg)的總離子流圖Fig.3 Total ion chromatogram of safrole in positive snack noodle(370 μg/kg)
2.6 實際樣品的測定
黃樟素屬天然香料,在許多天然植物中廣泛存在。采用本方法對市場上購買的姜、洋蔥、蔥、蒜、肉蔻、八角、桂皮、茴香、荊芥、石香、香菜、芹菜、調味料、方便面等46個樣品進行黃樟素、異黃樟素、二氫黃樟素的含量測定。結果表明,除蔥、洋蔥、石香、芹菜未檢出黃樟素外,其它樣品均檢出黃樟素,其中蒜芥、蒜、香菜含有少量黃樟素(<50 μg/kg),姜、桂皮和方便面中檢出黃樟素含量為228.1~731 μg/kg,茴香、肉蔻、八角和調味品中黃樟素含量為7.2~1 503.2 mg/kg。所有樣品均未檢出異黃樟素和二氫黃樟素。圖3為方便面中檢出黃樟素的總離子流圖。
3 結 論
本文建立了氣相色譜-質譜技術測定食品中黃樟素、異黃樟素、二氫黃樟素含量的分析方法。通過優化最佳的提取條件,聯用無水硫酸鈉、氟羅里硅土和PRiME HLB固相萃取的凈化方法,全掃描和選擇離子同時監測用于定量檢測和定性確證,驗證了18種不同的食品基質。方法參數均滿足《GB/T 27404-2008 實驗室質量控制規范 食品理化檢測》的要求,為食品中黃樟素及其衍生物的風險評估提供了可靠的技術基礎。
[1] Wang H L,Meng Z Y,Tang J G,Lu S M,Zhu R Z,Liu Z C,Liu C B.FlavourFragranceCosmetics(王海利,孟昭宇,湯建國,陸舍銘,朱瑞芝,劉正聰,劉春波.香料香精化妝品),2009,12(6):11-13.
[2] Stanfill S B,Ashley D L.J.Chromatogr.A,1999,858:79-89.
[3] Martati E,Boonpawa R,Berg J H,Paini A,Spenkelink A,Punt A,Vervoort J,Bladeren P J,Rietjens I M.FoodChem.Toxi.,2014,66(4):373-384.
[4] Shen L C,Chiang S Y,Lin M H,Chung W S,Wu K Y.Toxicol.Lett.,2012,213(9):309-315.
[5] Raffo A,Aloise A D,MagrìA L,Leclercq C.FoodChem.Toxicol.,2013,59(9):626-635.
[6] European Commission.Flavourings & Certain Food Ingredients.http://eur-lex.europa.eu/eli/reg/2008/1334/oj.
[7] Huang S S,Fan Z,Huang S J,Wang W G,Jiang H L,Zhu J,Lu B L.J.AnhuiAgric.Sci.(黃善松,范忠,黃世杰,王維剛,蔣宏霖,朱靜,陸冰琳.安徽農業科學),2014,42(20):6805-6806.
[8] Li Y,Wang H Y,Liao X L,Xiao S H,Chen H.Tobac.Sci.Technol.(李韻,汪宏毅,廖曉玲,肖少紅,陳慧.煙草科技),2014,47(12):32-35.
[9] Li C Y,Li Z G,Zhou S Y,Ye D F,Mo W M.Physi.Test.Chem.Anal.:Chem.Anal.(李長于,李祖光,周示玉,葉丹鳳,莫衛民.理化檢驗-化學分冊),2013,49(2):216-218.
[10] Wang X,Cai T P,Wang C,Xiao H Q,Zhang F,Liu L.J.Environ.Health(王星,蔡天培,王超,肖海清,張帆,劉柳.環境與健康雜志),2007,24(5):355-359.
[11] Zhu X L,Hong S Q,Li P P,Gao Y,Liu Y G.J.Instrum.Anal.(朱曉蘭,洪深求,李盼盼,高蕓,劉淵根.分析測試學報),2012,31(3):351-354.
[12] Stuppner H,Ganzera M.Chromatographia,1998,47(11/12):686-688.
[13] Zhou Y B,Teng M D,Zhang M S.Physi.Test.Chem.Anal.:Chem.Anal.(周貽兵,滕明德,張明時.理化檢驗-化學分冊),2011,47(1):39-40.
[14] Han Y,Zhang C M,Yu K,Meng X,Chen Y K,Miao M M.FlavourFragranceCosmetics(韓熠,張承明,喻坤,孟霞,陳永寬,繆明明.香料香精化妝品),2013,16(1):45-49.
[15] Siano F,Ghizzoni C,Gionfriddo F,Colombo E,Servillo L,Castaldoa D.FoodChem.,2003,81:469-475.
Determination of Safrole and its Derivatives in Foodstuffs by Gas Chromatography-Mass Spectrometry
ZHU Wei-xia1*,LI Sui2,ZHANG Li1,HU Kai2,YANG Ji-zhou1,ZHANG Shu-xia1
(1.Henan Entry-Exit Inspection and Quarantine Bureau,Zhengzhou 450003,China;2.College of Pharmacy,Henan University of Chinese Medicine,Zhengzhou 450000,China)
An analytical method was developed for the determination of safrole(SFL),isosafrole(DSFL) and dihydrosafrole(ISFL) in 18 foods including condiment,tomato,sauce,soy sauce,pepper,carbonated beverage,milk drink,juice,sausage,canned meat,meat soup,jelly,jam,dried fish,canned fish,fish slime,potato chip,candy and chocolate by gas chromatography-mass spectrometry(GC-MS).The sample was extracted with dichloromethane-acetonitrile in the presence of sodium chloride salt-out.Water and polar substances were removed with anhydrous sodium sulfite-florisil column.For complex matrices,further purification was performed with a PRiME HLB solid-phase extraction columns.The separation was carried out on a 5%phenyl-95% methyl polysiloxane capillary GC column.The detection of compounds was completed in the selected ion monitoring mode(SIM).The quantification analysis was performed by the internal standard method.Good linear relationships(r2≥0.998 1) betweeen peak areas ratios and concentrations ratios of 3 analytes were obtained in the range of 25-5 000 μg/kg.The limits of detection(LOD) for 3 analytes were 20 μg/kg.And the limits of quantitation(LOQ) were 50 μg/kg.The average recoveries of the method varied from 77.6% to 100.9% at 3 different spiked levels,with relative standard deviations(RSD) of 3.7%-13.6%.By measuring real samples,the results showed that this method was accurate and reliable for quantification and confirmation of SFL,DSFL and ISFL in foodstuffs.
gas chromatography-mass spectrometry(GC-MS);selected ion monitor(SIM);solid-phase extraction(SPE);safrole;spice additives;foodstuffs
2016-06-27;
2016-08-18
出入境檢驗檢疫行業標準制定項目(2012B137);國家質檢總局科技計劃項目(2015IK115)
10.3969/j.issn.1004-4957.2016.12.014
O657.63;Q946.85
A
1004-4957(2016)12-1596-05
*通訊作者:祝偉霞,碩士,高級工程師,研究方向:食品安全檢測,Tel:0371-55196533,E-mail:wxzhu121@163.com
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學生數理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
