2-(1-甲基-3-吡唑基)-5-溴吡啶的合成研究*
2017-01-16 05:28:12王昕悅倪治彬趙春深
廣州化工 2016年23期
王昕悅,倪治彬,溫 洋,楊 縉,趙春深
(貴州大學藥學院,貴州 貴陽 550025)
?
2-(1-甲基-3-吡唑基)-5-溴吡啶的合成研究*
王昕悅,倪治彬,溫 洋,楊 縉,趙春深
(貴州大學藥學院,貴州 貴陽 550025)
2-(1-甲基-3-吡唑基)-5-溴吡啶是一種重要的醫藥化工中間體。以2,5-二溴吡啶為起始原料,經取代、環合得到中間體5-溴-2-(1H-3-吡唑基)吡啶。進一步與碘甲烷發生取代反應,得到目標產物和副產物2-(2-甲基-3-吡唑基)-5-溴吡啶,結構經MS和1H NMR確認。本文考察了不同的縛酸劑對5-溴-2-(1H-3-吡唑基)吡啶取代反應的影響,確定了反應最佳條件,總收率為53.2%。該合成方法原料簡單易得,操作簡單,反應時間短且收率較高,適合工業化生產。
2,5-二溴吡啶;2-(1-甲基-3-吡唑基)-5-溴吡啶;2-(2-甲基-3-吡唑基)-5-溴吡啶;合成
雜環是現代農藥和醫藥領域新藥合成的重要特征結構,含吡唑環的化合物廣泛的應用于抗流感藥物、除草劑等醫藥有機農藥中[1-2]。吡啶是一種具有芳香性的含氮化合物,在醫藥、農藥、香料、染料等方面均有較廣泛的應用。溴代吡啶類化合物作為重要的含吡啶結構的中間體在農藥和醫藥領域也有著比較廣泛的用途,含溴吡啶類化合物不僅可以作為中間體直接在農藥和醫藥中應用,同時又是吡啶類含氟化合物合成的中間體原料,所以含溴吡啶化合物的合成在吡啶類化合物的合成中占非常重要的地位[3-4]。多重耐藥的革蘭氏陽性菌的數量隨著時間的推移迅速增加,2-(1-甲基-3-吡唑基)-5-溴吡啶(1A)是一種特效抗菌藥物[5-6]。2-(1-甲基-3-吡唑基)-5-溴吡啶上的溴和甲基的間位都比較容易引入其他官能團對結構進行修飾,能合成具有獨特作用機理的藥物,是合成mTOR抑制劑和PDE10抑制劑的重要中間體[7-8],有較好的市場應用前景。
目前,已報道的2-(1-甲基-3-吡唑基)-5-溴吡啶的合成方法是以5-溴-2-(1H-3-吡唑基)吡啶(4)為原料,選用KOH為縛酸劑反應得到[9],但該方法得到副產物2-(2-甲基-3-吡
唑基)-5-溴吡啶的比例較高,反應時間較長,且后處理需要柱層析,不適合工業化生產。本文選用2,5-二溴吡啶(2)為起始原料,先與N-甲氧基-N-甲基乙酰胺反應得到5-乙酰基-2-溴吡啶(3),再與DMF-DMA和水合肼經環合反應得到中間體5-溴-2-(1H-3-吡唑基)吡啶(4),中間體4與碘甲烷在縛酸劑作用下經取代反應得到目標化合物,為了得到收率和純度較高的產品,本文對比了不同縛酸劑對5-溴-2-(1H-3-吡唑基)吡啶取代反應的影響,探尋更好的反應條件。
目標化合物的具體合成路線如圖1所示。
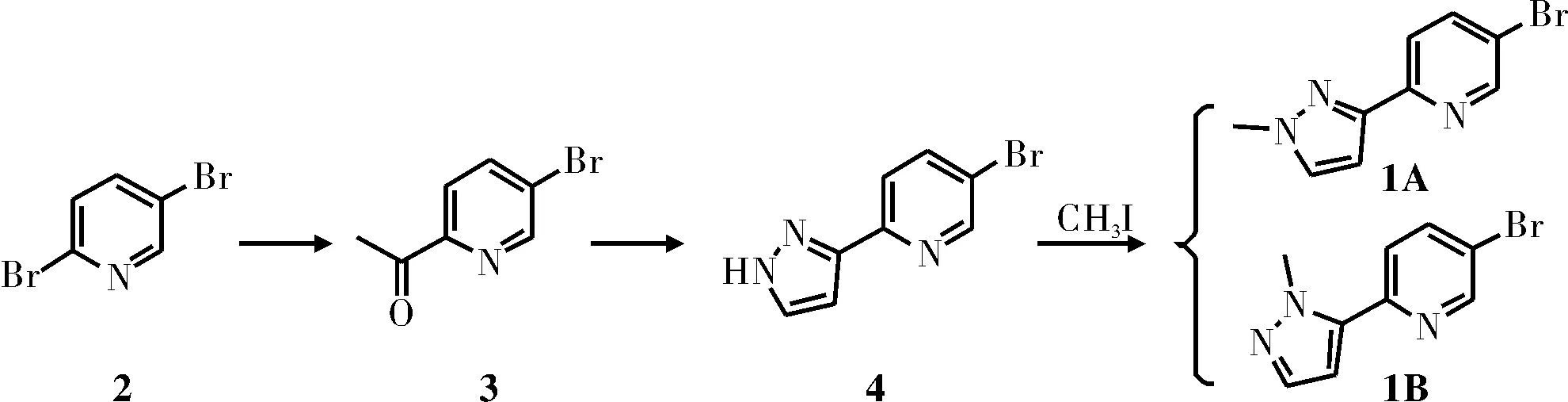
圖1 目標化合物合成路線
1 實 驗
1.1 主要儀器和試劑
DF-101S集熱式恒溫加熱磁力攪拌器,上海科升儀器有限公司;ZF7三用紫外分析儀,鞏義市予華儀器有限責任公司;DB30電子天平,上海臺衡儀器儀表有限公司;SHZ-型循環水真空泵,河南鞏義英峪儀器廠;Advance DMX400型核磁共振儀,Bruker公司(400 MHz,TMS為內標);Agilent 1100 LC-MS質譜,美國Agilent公司;N1200系列旋轉蒸發儀,東京理化器械株式會社;飛躍牌真空泵,上海禾汽玻璃儀器有限公司。
所用試劑均為市售化學純和分析純,并按要求進行純化處理。
1.2 制備方法
1.2.1 5-乙酰基-2-溴吡啶(3)的合成
在250 mL三口瓶中加入2,5-二溴吡啶(10.0 g,1 eq,42.2 mmol)和80 mL干燥的甲苯,氮氣保護,降溫至-40 ℃,控制溫度-40 ℃,緩慢滴加正丁基鋰(16.9 mL,1 eq,42.2 mmol),滴畢,在-40 ℃攪拌40 min,緩慢滴加N-甲氧基-N-甲基乙酰胺(7.9 g,1.8 eq,76.8 mmol)(控溫-40 ℃),滴畢,自然恢復至室溫,TLC檢測反應進度,反應完畢,用飽和氯化銨水溶液淬滅,乙酸乙酯(80 mL×3)萃取,合并有機相,用飽和食鹽水洗滌,無水硫酸鈉干燥,減壓蒸除溶劑,得7.1 g紅褐色固體,收率為84.1%。1H NMR (400 MHz, CDCl3) δ 8.88 (s, 1H), 8.05 (dd,J=9.4, 3.6 Hz, 1H), 7.59 (d,J=8.3 Hz, 1H), 2.60 (s, 3H)。
1.2.2 5-溴-2-(1H-3-吡唑基)吡啶(4)的合成
在100 mL單口瓶中加入5-乙酰基-2-溴吡啶(4.0 g,1 eq,20.0 mmol)和DMF-DMA(11.9 g,5 eq,100.0 mmol),升溫至回流,攪拌5 h,反應完畢,冷卻至室溫,蒸除溶劑,得到紅褐色固體。將紅褐色固體溶于40 mL乙醇中,加入水合肼(3.0 g,3 eq,60.0 mmol),升溫至回流,攪拌2 h,反應完畢,冷卻至室溫,蒸除溶劑,得到黃褐色固體。用少量甲醇打漿,抽濾,干燥得4.2 g淡黃色固體產品,收率為85.1%。1H NMR (400 MHz, CDCl3) δ 8.70 (dd,J=2.3, 0.5 Hz, 1H), 7.86 (dd,J=8.4, 2.3 Hz, 1H), 7.68 (dd,J=7.3, 5.3 Hz, 2H), 6.82 (d,J=2.1 Hz, 1H)。
1.2.3 2-(1-甲基-3-吡唑基)-5-溴吡啶(1A)的合成
在100 mL三口瓶中加入5-溴-2-(1H-3-吡唑基)吡啶(2.0 g,1 eq,8.9 mmol)和氫化鈉(含量60%,2.5 g,3 eq,26.7 mmol),加入DMF 20 mL溶解,冰浴下緩慢滴加碘甲烷(5.1 g, 4 eq, 35.6 mmol)的5 mL DMF溶液,滴畢,保持0 ℃繼續攪拌1 h。反應完成后,加入80 mL水淬滅,用乙酸乙酯(15 mL×3)萃取,合并有機相,用飽和食鹽水洗滌,無水硫酸鈉干燥,減壓蒸除溶劑得固體粗品1.8 g,用少量乙醚打漿,抽濾,干燥得純品(1A) 1.5 g,收率為74.4%。MS, m/z: 238.1[M+H]+;1H NMR (400 MHz, CDCl3) δ 8.70 (s, 1H), 7.84 (dd,J=8.4, 2.3 Hz, 1H), 7.47 (dd,J=9.6, 5.2 Hz, 2H), 6.56 (d,J=1.9 Hz, 1H), 4.18 (s, 3H)。
2 結果與討論
本文對比了不同縛酸劑對5-溴-2-(1H-3-吡唑基)吡啶取代反應的影響,結果如表1所示。
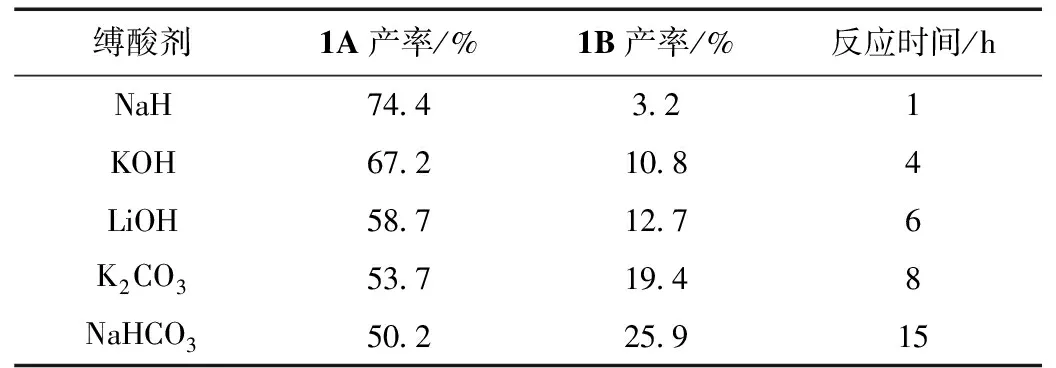
表1 縛酸劑對5-溴-2-(1H-3-吡唑基)吡啶取代反應的影響
實驗結果表明,當選用氫化納做為縛酸劑時,副產物的含量為3.2%,反應時間為1 h;氫氧化鉀做為縛酸劑時,副產物的含量為10.8%,反應時間為4 h;氫氧化鋰為縛酸劑時副產物的含量為12.7%,反應時間為6 h;碳酸鉀為縛酸劑時副產物的含量為19.4%,反應時間為8 h;碳酸氫鈉為縛酸劑時副產物的含量為25.9%,反應時間為15 h。因此,對比不同縛酸劑的反應效果可知,選用NaH做縛酸劑,不僅能減少副產物,縮短反應時間,產品的后處理簡單且收率較高,適合工業化生產。
[1] 黃長江,劉冰妮,張士俊,等.吡唑類化合物的合成與藥理活性研究[J].中國新藥雜志,2007,16(24):2043-2044.
[2] 任雪玲,胡方中,鄒小毛,等.吡唑聯吡唑化合物庫的液相平行合成[J].農藥,2003,42(6):17-18.
[3] 孫蔭輝.吡啶類化合物的合成與應用[J].江蘇化工,1996(5):9-14.
[4] 徐杰,張偉.鹵代吡啶類化合物的合成及應用[J].精細化工原料及中間體,2006,36(4):1-6.
[5] 劉浚,孟慶國,金潔,等.噁唑烷酮類抗菌劑研究進展[A].2001中國藥學會學術年會[C].2001.
[6] 楊燕,尤啟冬.噁唑烷酮類抗菌劑構效關系及結構改造研究進展[J].藥學進展,2010,34(11):481-490.
[7] Siddiqui M A, Nan Y, Patel M F, et al. Pyrazolo[1,5-a]pyimidine compounds as mTOR inhibitors[P]. WO2011090935.
[8] Cai Z, Zhou D, LIN Y, et al. Alkyne-bridged hetero-aromatics and uses thereof [P]. WO 20130158031.
[9] Jo Y W, Im W B, Rhee J K, et al. Synthesis and antibacterial activity of oxazolidinones containing pyridine substituted with heteroaromatic ring[J]. Bioorganic & medicinal chemistry, 2004, 12(22): 5909-5915.
Synthesis of 2-(1-methyl-3-imidazolyl)-5-bromopyridine*
WANGXin-yue,NIZhi-bin,WENYang,YANGJin,ZHAOChun-shen
(Guizhou University, School of Pharmaceutical Sciences, Guizhou Guiyang 550025, China)
2-(1-methyl-3-pyrazolyl)-5-bromo-pyridine is an important pharmaceutical and chemical intermediate. Taking 2,5-dibromopyridine as a starting material, after substituted halogen, addition and cyclization, the intermediate 5-bromo-2-(1H-3- pyrazolyl)pyridine was synthesized. After further substitution reaction with methyl iodide, the desired product and by-product 2-(2-methyl-3-imidazolyl)-5-bromopyridine were synthesized, their structures were confirmed by MS and1H NMR. The influence of different acid-binding agents on the substitution of 5-bromo-2-(1H-3-pyrazolyl)pyridine was investigated. The optimum reaction conditions were determined, total yield was 53.2%. The synthesis method is simple and easy to obtain, simple in operation, short in reaction time and high in yield, and is suitable for industrial production.
2,5-dibromo-pyridine; 2-(1-methyl-3-pyrazolyl)-5-bromopyridine; 2-(2-methyl-3-pyrazolyl)-5-bromopyridine; synthesis
貴州大學“大學生創新創業訓練計劃”項目,合同編號:貴大(國)創字2014(022)。
王昕悅(1995-),女,制藥工程專業本科生。
O626.3
A
1001-9677(2016)023-0044-02
