(4-(N-環(huán)丙基氨磺酰)-2,5-二氟苯基)硼酸的合成*
2017-01-16 05:28:12楊紅梅趙春深
廣州化工 2016年23期
楊紅梅,翁 崢,姚 燕,趙春深
(1 貴州大學(xué)藥學(xué)院,貴州 貴陽 550003;2 貴州省發(fā)酵工程與生物制藥重點實驗室,貴州 貴陽 550003)
?
(4-(N-環(huán)丙基氨磺酰)-2,5-二氟苯基)硼酸的合成*
楊紅梅1,2,翁 崢1,2,姚 燕1,2,趙春深1,2
(1 貴州大學(xué)藥學(xué)院,貴州 貴陽 550003;2 貴州省發(fā)酵工程與生物制藥重點實驗室,貴州 貴陽 550003)
(4-(N-環(huán)丙基氨磺酰)-2,5-二氟苯基)硼酸是一個重要的化工中間體,目前無合成路線報道。通過查閱文獻制定了最佳合成路線,本文設(shè)計了一條以2,5-二氟溴苯為起始原料,經(jīng)磺酰化、酰胺化和取代反應(yīng)得到目標(biāo)化合物的合成路線。通過大量平行實驗,對溫度、投料比例等實驗因素進行考察,得出了最佳反應(yīng)條件。該方法原料廉價易得,方法簡單,操作簡便,收率較高,適合工業(yè)化生產(chǎn)。
2,5-二氟溴苯;(4-(N-環(huán)丙基氨磺酰)-2,5-二氟苯基)硼酸;合成
硼酸類化合物廣泛應(yīng)用于玻璃、塘瓷、陶瓷和輕紡織等工業(yè)領(lǐng)域[1]。因其弱酸溫和性,硼酸化合物在醫(yī)藥行業(yè)也得到了廣泛的應(yīng)用[2]。苯硼酸化合物作為硼酸類化合物的重要分支,可以與多種羥基化合物形成可逆化合物,在自律式胰島素給藥系統(tǒng)、生物物質(zhì)分離系統(tǒng)以及傳感器方面均有應(yīng)用,具有廣泛的應(yīng)用前景[3]。(4-(N-環(huán)丙基氨磺酰)-2,5-二氟苯基)硼酸(4)是合成酪氨酸激酶抑制劑的重要中間體[4]。
目前,(4-(N-環(huán)丙基氨磺酰)-2,5-二氟苯基)硼酸的合成未見文獻報道。本文對其合成路線進行了設(shè)計,以2,5-二氟溴苯(1)為起始原料,先與氯磺酸磺酰化反應(yīng)得到4-溴-2,5-二氟苯磺酰氯(2),再與環(huán)丙胺酰胺化反應(yīng)得到4-溴-N-環(huán)丙基-2,5-二氟苯磺酰胺(3),最后與硼酸三異丙酯和正丁基鋰反應(yīng)得到目標(biāo)化合物。
1 合成路線對比
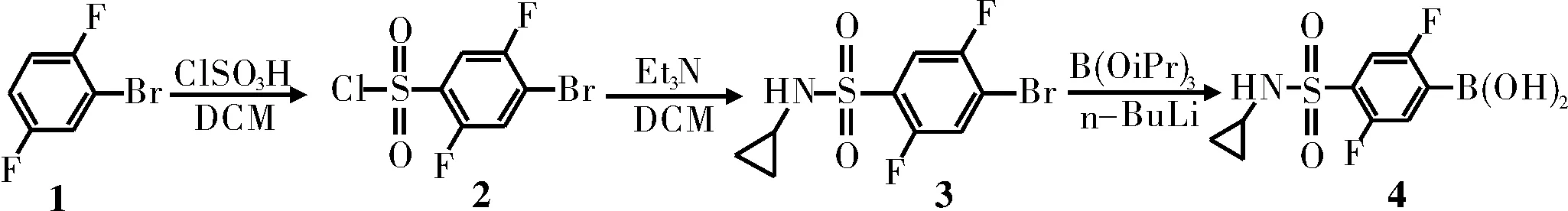
圖1 合成路線1
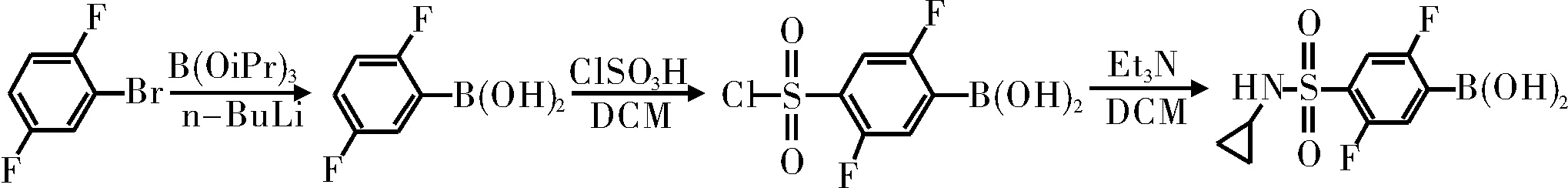
圖2 合成路線2
通過對比合成路線1和合成路線2,因為硼酸不穩(wěn)定,遇強酸易變質(zhì),所以為了避免硼酸變質(zhì),將取代反應(yīng)放在最后一步,選擇合成路線1。
2 實 驗
2.1 主要儀器與試劑
DF-101S集熱式恒溫加熱磁力攪拌器,鞏義市予華儀器有限責(zé)任公司;三用紫外分析儀,上海市安亭電子儀器廠;DB30電子天平,鞏義市予華儀器有限責(zé)任公司;Advance DMX400型核磁共振儀(400 MHz,TMS為內(nèi)標(biāo));YRE2000B旋轉(zhuǎn)蒸發(fā)儀,鞏義市予華儀器有限責(zé)任公司。
所用試劑均為市場出售化學(xué)純和分析純,并按要求進行純化處理。
2.2 制備方法
2.2.1 4-溴-2,5-二氟苯磺酰氯(2)的制備
冰浴下,在500 mL三口瓶中加入2,5-二氟溴苯(30.00 g,125.00 mmol,1.00 eq),緩慢滴加用二氯甲烷稀釋的氯磺酸(31.80 g, 274.00 mmol,2.20 eq),滴畢,升至室溫攪拌1 h,反應(yīng)完畢后,將反應(yīng)液倒入冰水中,用二氯甲烷(200 mL×2)萃取,合并有機相,用無水硫酸鈉干燥,蒸除溶劑得淡黃色固體39.70 g,收率為87.64%。

表1 15 ℃時不同投料比的反應(yīng)情況
由表1可得出,溫度在5 ℃不變的情況下,在投料比為1:1和1:1.5時,反應(yīng)時間為5 h的情況下,大量未反應(yīng)。在投料比為1:2,反應(yīng)時間為2 h的情況下,反應(yīng)完全。在投料比為1:2.2,反應(yīng)時間為1 h的情況下,反應(yīng)完全。工業(yè)生產(chǎn)考慮,為了降低生產(chǎn)成本。所以選擇1:2的投料比。
2.2.2 4-溴-N-環(huán)丙基-2,5-二氟苯磺酰胺(3)的制備
在500 mL三口瓶中,加入4-溴-2,5-二氟苯磺酰氯(20.00 g,69.00 mmol,1.00 eq)和200 mL二氯甲烷,冰浴下,滴加環(huán)丙胺(4.7 g,82.5 mmol,1.20 eq)的二氯甲烷溶液,滴畢,升至室溫攪拌3 h,反應(yīng)完畢后,結(jié)束后,將反應(yīng)液倒入水中,用二氯甲烷(150 mL×2)萃取,合并有機相,用無水硫酸鈉干燥,蒸除溶劑得黃色固體20.71 g,收率為96.70%。
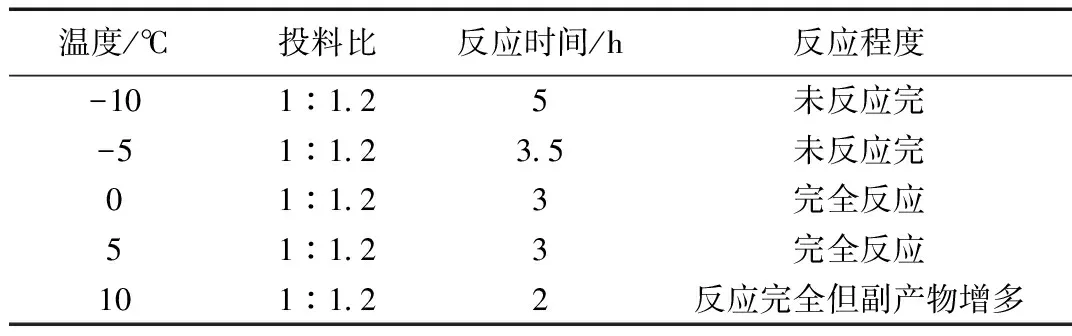
表2 不同溫度的反應(yīng)情況
由表2可得出,投料比1:1.2不變的情況下,在溫度在-10 ℃,反應(yīng)時間為5 h的情況下,未反應(yīng)完。在溫度為-5 ℃,反應(yīng)時間為3.5 h的情況下,未反應(yīng)完,在溫度為0~5 ℃,反應(yīng)時間為3 h的情況下,完全反應(yīng)。在溫度為10 ℃,反應(yīng)時間2 h的情況下,反應(yīng)完全但副產(chǎn)物增多。綜上所述,選擇冰浴溫度為0~5 ℃。
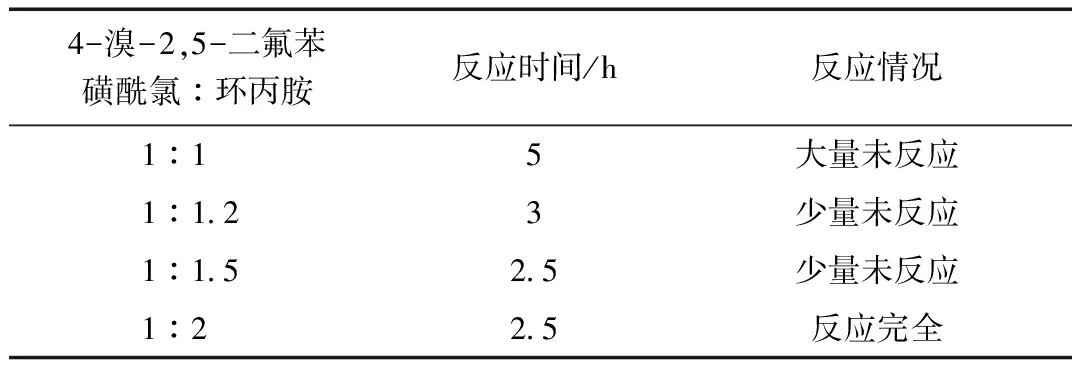
表3 0~5 ℃時不同投料比的反應(yīng)情況
由表3可得,在溫度為0~5 ℃時,投料比為1:1,反應(yīng)時間5 h時,大量未反應(yīng)。當(dāng)投料比為1:1.2,反應(yīng)時間為3 h時,少量未反應(yīng)。當(dāng)投料比為1:1.5,反應(yīng)時間為2.5 h時,少量未反應(yīng)。當(dāng)投料比為1:2,反應(yīng)時間為2.5 h時,反應(yīng)完全。綜上所述,選擇投料比為1:1.2。2.2.3 (4-(N-環(huán)丙基氨磺酰)-2,5-二氟苯基)硼酸(4)的制備
在250 mL三口瓶中,加入 4-溴-N-環(huán)丙基-2,5-二氟苯磺酰胺(10.00 g, 32.00 mmol,1.00 eq)、硼酸三異丙酯(7.20 mL,38.40 mmol,1.20 eq)和50 mL無水2-甲基四氫呋喃。N2保護下,降溫至-78 ℃以下,緩慢滴加正丁基鋰(28.00 mL, 70.40 mmol,2.20 eq),滴畢,在-78 ℃下攪拌30 min后恢復(fù)至室溫攪拌1 h,反應(yīng)完畢,緩慢加水淬滅反應(yīng),用乙酸乙酯(50 mL×3)萃取,合并有機相,用無水硫酸鈉干燥,蒸除溶劑得白色固體粗品8.02 g,柱層析(石油醚:乙酸乙酯=5:1),得到純品6.47 g,產(chǎn)率為72.88%。MS:278.04 [M+H]+,1H NMR (400 MHz, DMSO) δ 8.38 (d, J=2.4 Hz, 1H), 7.70~7.44 (m, 3H), 2.26 (dd, J=6.7, 3.4 Hz, 1H), 0.52~0.46 (m, 2H), 0.45~0.30 (m, 2H)。
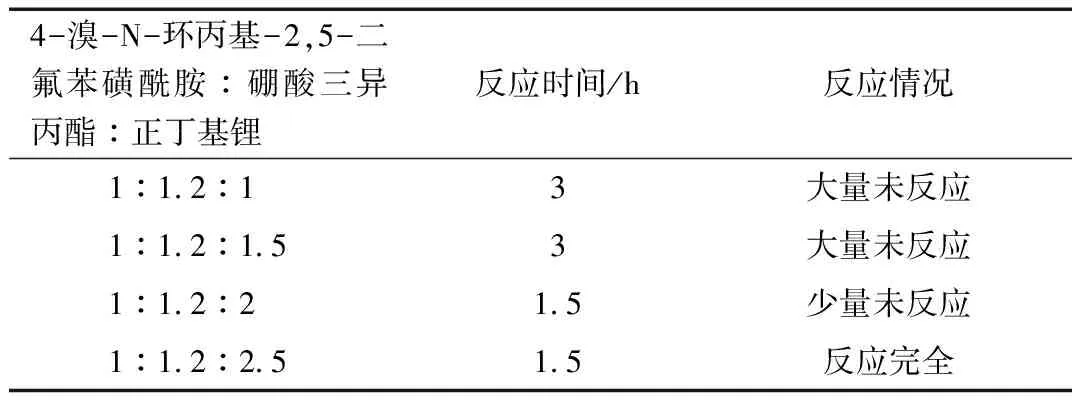
表4 -78 ℃時不同投料比的反應(yīng)情況
由表4可得,在溫度為-78 ℃時,投料比為1:1.2:1,反應(yīng)時間3 h時,大量未反應(yīng)。當(dāng)投料比為1:1.2:1.5,反應(yīng)時間為3 h時,大量未反應(yīng)。當(dāng)投料比為1:1.2:2,反應(yīng)時間為1.5 h時,少量未反應(yīng)。當(dāng)投料比為1:1.2:2.5,反應(yīng)時間為1.5 h時,反應(yīng)完全。綜上所述,選擇投料比為1:1.2:2.5。
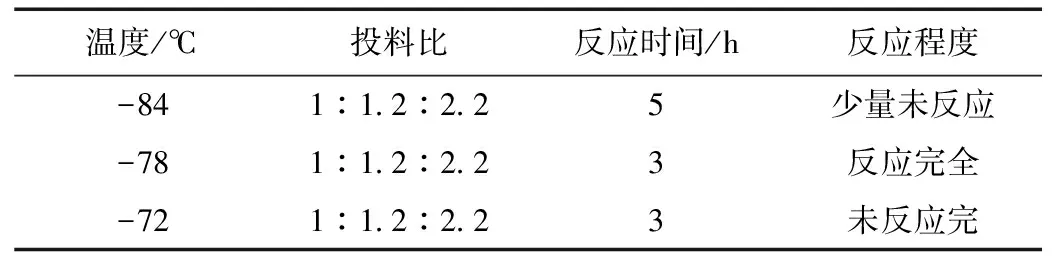
表5 投料比為1:1.2:2.2時不同溫度的反應(yīng)情況
由表5可得出,投料比1:1.2:2.2不變的情況下,在溫度在-84 ℃,反應(yīng)時間為5 h的情況下,少量未反應(yīng)。在溫度為-78 ℃,反應(yīng)時間為3 h的情況下,反應(yīng)完全,在溫度為-72 ℃,反應(yīng)時間為3 h的情況下,未反應(yīng)完。綜上所述,選擇溫度為-78 ℃。
3 結(jié) 論
本文提供了(4-(N-環(huán)丙基氨磺酰)-2,5-二氟苯基)硼酸的合成路線,以2,5-二氟溴苯為起始原料,經(jīng)磺酰化、酰胺化和取代反應(yīng)得到目標(biāo)化合物的合成路線,確定了磺酰化反應(yīng)最佳投料比為2,5-二氟溴苯:氯磺酸為1:2.2,酰胺化反應(yīng)的最佳反應(yīng)條件:投料比為4-溴-2,5-二氟苯磺酰氯:環(huán)丙胺為1:1.2,溫度為0~5 ℃;取代反應(yīng)的最佳反應(yīng)條件為:投料比為4-溴-N-環(huán)丙基-2,5-二氟苯磺酰胺:硼酸三異丙酯:正丁基鋰為1:1.2:2.2,反應(yīng)溫度為-78 ℃。該方法原料廉價易得,方法簡單,操作簡便,總收率為61.76%,適合工業(yè)化生產(chǎn)。
[1] 龔殿婷,李鳳華,劉素蘭,等. 硼酸的生產(chǎn)應(yīng)用現(xiàn)狀及市場前景[J]. 化學(xué)工業(yè)與工程技術(shù), 2007, 28(6):50-54.
[2] 徐丹, 褚良銀. 苯硼酸及其衍生物在醫(yī)藥與化工領(lǐng)域的應(yīng)用研究進展[J]. 化工進展, 2006, 25(9):1045-1048.
[3] 周公度. 硼酸鹽的結(jié)構(gòu)化學(xué)[J]. 化學(xué)通報,1960(3):113-121
[4] Ali A M, Gómez-Biagi R F, Rosa D A, et al. Disarming an Electrophilic Warhead: Retaining Potency in Tyrosine Kinase Inhibitor (TKI)-Resistant CML Lines While Circumventing Pharmacokinetic Liabilities[J]. ChemMedChem, 2016, 11(8): 850-861.
[5] Foschi F, Tagliabue A, Mihali V, et al. Memory of chirality approach to the enantiodivergent synthesis of chiral benzo [d] sultams[J]. Organic letters, 2013, 15(14): 3686-3689.
Synthesis of(4-(N-cyclopropylsulfamoyl)-2,5-difluorophenyl)boronic Acid*
YANGHong-mei1,2,WENGZheng1,2,YAOYan1,2,ZHAOChun-shen1,2
(1 College of Pharmacy, Guizhou University, Guizhou Guiyang 550003; 2 Key Laboratory of Fermentation Engineering and Biopharmaceutics of Guizhou Province, Guizhou Guiyang 550003, China)
(4-(N-cyclopropylsulfamoyl)-2,5-difluorophenyl)boronic acid is an important pharmaceutical chemical intermediates, there is no synthesis route reported. The synthesis route of the target compound was obtained by the sulfonylation, amidation and substitution of 2,5-difluorobromobenzene as starting materials. Through a large number of parallel experiments, the temperature, feed ratio and other experimental factors were investigated, the best reaction conditions were obtained. The method has the advantages of low cost, simple method, simple operation and high yield, and is suitable for industrial production.
2,5-difluorobromobenzene; 4-bromo-N-cyclopropyl(4-(N-cyclopropylsulfamoyl)-2,5-difluorophenyl)boronic acid; synthesis
貴州大學(xué)大學(xué)生“SRT計劃”項目[合同編號:貴大SRT字(2015)077號]。
楊紅梅(1997-),女,藥物制劑專業(yè)本科生。
趙春深(1978-),男,副教授,主要從事藥物合成研究。
O625.66
A
1001-9677(2016)023-0046-03
