微波輔助、磷酸催化合成半乳聚糖的結構分析
2017-03-21 10:19:08王海松丁寅翼成向榮樂國偉
食品與生物技術學報 2017年1期
關鍵詞:信號
紀 高, 王海松, 丁寅翼, 成向榮*,2, 樂國偉,2
微波輔助、磷酸催化合成半乳聚糖的結構分析
紀 高1, 王海松1, 丁寅翼1, 成向榮*1,2, 樂國偉1,2
(1.江南大學 食品學院,江蘇 無錫 214122;2.食品科學與技術國家重點實驗室,江南大學,江蘇 無錫214122)
以磷酸催化、微波輔助合成半乳聚糖為研究對象,探討半乳聚糖的化學結構及聚合反應的區域選擇性和立體選擇性。綜合采用高效凝膠滲透色譜、紅外光譜、甲基化分析及一維、二維核磁共振波譜(1H NMR,13C NMR,1H-1H COSY,TOCSY,HSQC,HMBC),對半乳聚糖的相對分子質量、糖苷鍵的立體構型和連接位點進行表征。結果表明,反應合成的半乳聚糖為多分支結構,重均相對分子質量為2 657,重均聚合度為16,糖殘基以α-型半乳吡喃糖為主,主要為→4)-α-D-Galp(1→和→2,4)-α-D-Galp(1→片段。結構分析結果進一步表明,微波輔助、磷酸催化半乳糖縮合過程中,不同位點的羥基具有不同的反應活性,4位羥基的反應活性最強。
微波;磷酸;半乳聚糖;結構分析
半乳聚糖是一類廣泛存在于海洋藻類、陸生植 物或動物組織中,由半乳糖或以半乳糖為主、其它單糖為輔構成的直鏈或支鏈聚合物[1]。半乳聚糖,尤其是來源于藻類的半乳聚糖硫酸酯,具有增強免疫、抗病毒、抗凝血及抑制腫瘤轉移的生物學功能[2-5]。此外,半乳聚糖作為一類膳食纖維,還可以有效促進腸道乳酸菌增殖,提高腸道短鏈脂肪酸的含量[6]。
目前,半乳聚糖的制備主要依賴于天然資源的提取、水解和衍生。天然資源的生長周期性和提取工藝的復雜性嚴重制約了半乳聚糖的深入開發和應用。結合糖在酸催化、加熱條件下可脫水縮合的特點,以及微波輻射具有加熱均勻、快速的優勢,作者研究了微波輔助快速合成低聚糖、多糖的技術,并分別以葡萄糖、甘露糖、半乳糖為原料,在雜多酸催化下,快速、高收率地合成了相應低聚糖或多糖[7-9]。糖類物質的化學結構中普遍存在多個位點的異構,包括區域異構及立體異構,即糖苷鍵不同連接位置、不同立體化學的異構形式,是化學法合成低聚糖、多糖過程中面臨的主要挑戰[10-12],也是微波輔助合成聚糖過程中亟待解決的科學問題。若能夠掌握微波輻照、酸催化單糖聚合過程中的區域選擇性和立體選擇性規律,那么,微波輔助定向合成低聚糖、多糖將有可能成為現實。
王海松等對微波輻照、雜多酸催化半乳糖聚合的反應進行了優化,并對反應產物進行了初步的化學表征,結果發現合成的半乳聚糖平均聚合度為17,半乳糖殘基以β-構型為主[9],但糖苷鍵的連接位置尚未闡明。作者在上述優化條件的基礎上,以半乳糖為原料,采用食品中常用的酸化劑——磷酸為催化劑,微波輻照合成半乳聚糖,并對合成產物進行細致的結構分析,總結半乳糖聚合過程中的區域選擇性和立體選擇性。
1 材料與方法
1.1 材料與試劑
D-半乳糖(Gal)、氯化鈉、磷酸、三氟乙酸、無水乙醇(99%):國藥集團化學試劑有限公司產品;重水(D2O):上海晶純生化科技股份有限公司產品;Sephadex G-25葡聚糖凝膠:北京索萊寶科技有限公司產品。
1.2 儀器與設備
XH-200A型電腦微波固液相合成/萃取工作站:北京祥鵠科技發展有限公司產品;BSZ-100型自動部分收集器:上海青浦滬西儀器廠產品;R-205型旋轉蒸發儀:無錫申科公司產品;1260型高效液相色譜儀 (配Shodex RI-101示差檢測器):美國Agilent公司產品;ICS-5000型離子色譜儀器:美國Dionex公司產品;600型高效凝膠滲透色譜儀 (配2410視差檢測器):美國Waters公司產品;7890A-5975C型氣相色譜質譜聯用儀:美國安捷倫公司產品;560傅里葉變換紅外光譜儀:美國Nicolet公司產品;DRX-500核磁共振儀:德國Bruker公司產品。
1.3 方法
1.3.1 半乳聚糖的合成和純化 半乳聚糖的合成和純化參考王海松等的方法[9]。25 g D-半乳糖與1.1%磷酸、3.75 mL氯化鈉溶液(0.25 mol/L)充分攪拌混勻后送入微波反應器。設定微波功率800 W、反應溫度130℃、反應時間4.5 min,開啟攪拌器120 r/min,并開啟微波合成儀。反應結束后,得淡黃色膠狀固體。反應產物溶于去離子水中,經5倍體積的無水乙醇沉淀后,棄上清液。多糖沉淀加去離子水復溶,采用Sephadex G-25葡聚糖凝膠色譜柱分離,以去離子水作洗脫劑,流量0.5 mL/min,用部分收集器收集洗脫液,每管收集量為3 mL,時間間隔6 min。收集液中的糖組分采用硫酸-苯酚法測定[13],檢測波長為490 nm,以吸光度為縱坐標,繪制洗脫曲線。收集各管洗脫液,旋轉蒸發濃縮后,凍干得白色粉末狀樣品。
1.3.2 相對分子質量測定 采用高效凝膠滲透色譜法測定半乳聚糖的相對分子質量。標準品和樣品溶液:標準葡聚糖Dextran(相對分子質量為828,2 700,9 750,36 800,13 5350,Sigma-Aldrich公司產品);葡萄糖:Sigma-Aldrich公司產品;用流動相配成質量濃度為5 g/L的溶液,然后于5 000 r/min速率下離心10 min,取上清;色譜柱:TSKgel G3000 PWXL凝膠色譜柱(300 mm×7.8 mm,日本TOSOH公司);流動相:0.1 mol/L NaNO2含0.5 g/L NaN3;流量:0.9 mL/min;柱溫:45℃;進樣量:20 μL。將系列標準葡聚糖和葡萄糖相對分子質量的對數Lg Mw對其相應的保留時間tR進行回歸處理,得出線性回歸方程。
1.3.3 半乳聚糖紅外光譜分析 稱取半乳聚糖樣品1.3 mg,加入200目的KBr粉末150 mg,于紅外燈照射下,在瑪瑙乳缽中研磨均勻,裝入壓片磨具,用油壓機以20 MPa的壓力壓制2 min,然后用鑷子小心取下壓片,裝入樣品架,用Nicolet 560傅里葉變換紅外光譜儀于500~4 000 cm-1范圍內掃描紅外光譜。
1.3.4 甲基化分析 半乳聚糖的甲基化分析參考Hakomori的方法[14]。20 mg干燥的半乳聚糖樣品溶解于6 mL DMSO中,室溫振蕩2 h。在氬氣保護下加入甲基亞磺酰負離子(30 mg的NaH固體溶解于3 mL無水DMSO中,溫度控制在50~55℃,攪拌至反應液呈墨綠色,得甲基亞磺酰負離子),常溫下磁力攪拌至顏色退去,冰浴后緩慢加入4 mL碘甲烷,至溶液逐漸變為淡黃色。反應結束后加入少量蒸餾水終止反應,然后將生成物透析,冷凍干燥得甲基化半乳聚糖。向上述甲基化半乳聚糖中加入1 mL甲酸,110℃水解6 h,加熱除去甲酸,再加入2 mL濃度為2 mol/L的三氟乙酸,置于安瓿瓶中,充惰性氣體密封后100℃反應6 h,氬氣流中冷卻、干燥。在水解產物中加0.6 mL去離子水和50 mg NaBH4,室溫下過夜,加少量甲酸至無氣泡,加甲醇蒸干。然后加入1 mL的吡啶-乙酸酐(體積比為1∶1),100℃反應2 h,旋蒸干燥后加氯仿溶解,進行GC-MS分析,進樣量為1~2 μL。GC-MS分析色譜柱:Agilent DB-5ms(30 m×0.25 mm×0.25 μm);載氣流量(氦氣>99.99%):1.0 mL/min,分流比1∶50;程序升溫:80℃,保留5 min,以15℃/min升至200℃保留l min,再以10℃/min升至260℃,保留10 min;接口溫度250℃;EI+源:70 eV,250℃,掃描頻率5次/秒,質量范圍:33~600 amu。數據分析采用AMSD化學工作站軟件(version E.02.02.1431)和NIST11數據庫。
1.3.5 核磁共振波譜分析 半乳聚糖樣品前處理參考Hu等的方法[15]。25 mg半乳聚糖溶解于0.4 mL重水中,振蕩15 min,溶解后凍干,該步驟重復操作3次。最后將凍干的半乳聚糖樣品溶解在0.4 mL重水中,Bruker DRX-500核磁共振光譜儀檢測,記錄1H-NMR,13C-NMR,1H-1H COSY,TOCSY,HMBC,HSQC譜。1H-NMR和13C-NMR光譜測定頻率分別為500.13和125.77 MHz,樣品測定溫度65℃。
2 結果與討論
2.1 半乳聚糖的制備
中強酸可以促進糖苷鍵原子的質子化,加快多糖的水解和單糖的縮合。王海松等[9]采用微波輔助、雜多酸催化方法快速合成了半乳聚糖,并對合成工藝進行了優化。在本實驗中,根據優化后的反應條件,采用食品工業中常用的磷酸代替雜多酸來催化半乳糖的聚合。反應產物經過除雜、柱色譜分離,得到半乳聚糖22.6 g,反應得率為90.4%,與文獻[9]結果相近。實驗結果表明,磷酸能夠快速催化半乳糖聚合,與雜多酸相比,磷酸具有引入雜原子更少、反應產物更易處理的優勢。
2.2 相對分子質量測定
根據系列標準葡聚糖、葡萄糖溶液的保留時間,以保留時間tR對相對分子質量的對數lg Mw作圖,結果見圖1。多糖相對分子質量在180~135 350范圍內,Lg Mw與tR呈良好線性關系,回歸方程為Lg Mw=0.437 7tR+12.368,R2=0.998 8。
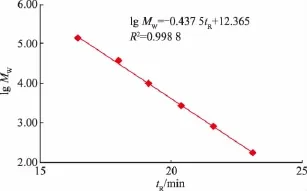
圖1 葡萄糖和葡聚糖標準曲線Fig.1 Calibration curve of glucose and dextrans
經過乙醇沉淀和柱色譜分離純化后,半乳聚糖的高效凝膠滲透色譜行為如圖2所示,為單一、狹窄、對稱的色譜峰,表明制備得到的半乳聚糖純度較高。分析結果表明,半乳聚糖的重均相對分子質量(Mw)為2 657,數均相對分子質量(Mn)為2 215,峰位相對分子質量(Mp)為2 196,重均聚合度為16,分子質量分布系數(HI=Mw/Mn)為1.19。HI值趨近于1,表明分離純化后的半乳聚糖分子質量的多分散程度小,合成制備的半乳聚糖分子質量分布較為均一。
2.3 紅外光譜分析
實驗合成的半乳聚糖和D-半乳糖的紅外光譜圖如圖3所示。與D-半乳糖相比,半乳聚糖位于3 000~3 700 cm-1處的O—H伸縮振動吸收峰較為寬鈍,提示半乳聚糖中存在豐富的、不同化學環境的羥基基團。王海松等認為1 647 cm-1處是C=O振動吸收峰[9],但C=O振動一般出現在1 725~1 730 cm-1處,這與紅外光譜信號并不一致,并且在1H和13C NMR圖譜(圖4)中未觀察到與羰基或羧基相對應的化學位移信號。水的特征吸收峰出現在3 420 cm-1和1 630 cm-1附近,故而推測1 647 cm-1處是水的吸收峰,表明半乳聚糖中存在微量的水分。D-半乳糖和半乳聚糖在1 000~1 200 cm-1處存在3個強吸收峰,表明兩者均為吡喃型糖苷。877 cm-1和793 cm-1處的吸收峰表明半乳聚糖中存在α-和β-構型的半乳糖殘基[9]。
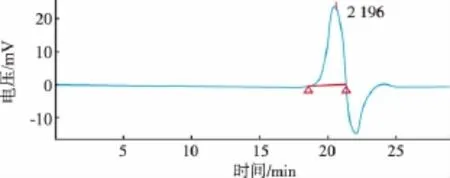
圖2 半乳聚糖的高效凝膠滲透色譜分析Fig.2 High performance gel permeation chromatography of galactan

圖3 微波輔助合成半乳聚糖和D-半乳糖的紅外光譜圖Fig.3 FT-IR spectra of synthesized galactan and galactose
2.4 甲基化分析
從半乳聚糖的甲基化結果(表1)可以看出,該多糖中半乳糖的主要連接方式為1,4和1,2,4連接,比例約為1.5∶1,而文獻報道紅藻來源的半乳聚糖的主鏈以1,3和1,4連接為主[16-17]。此外,微波輔助合成的半乳聚糖結構中還存在部分的1,6連接型半乳糖殘基,以及少量的1,3和1,4,6連接的半乳糖結構片段,而1,2和1,3,6連接的半乳糖殘基含量極低。這些結果表明,在反應過程中,半乳糖上不同位置羥基的反應活性不同,4-OH>6-OH>3-OH>2-OH。2,3,4,6-Me4-Gal為半乳糖末端,含量為4.2%,表明半乳聚糖具有較高的聚合度,與相對分子質量的測定結果一致。
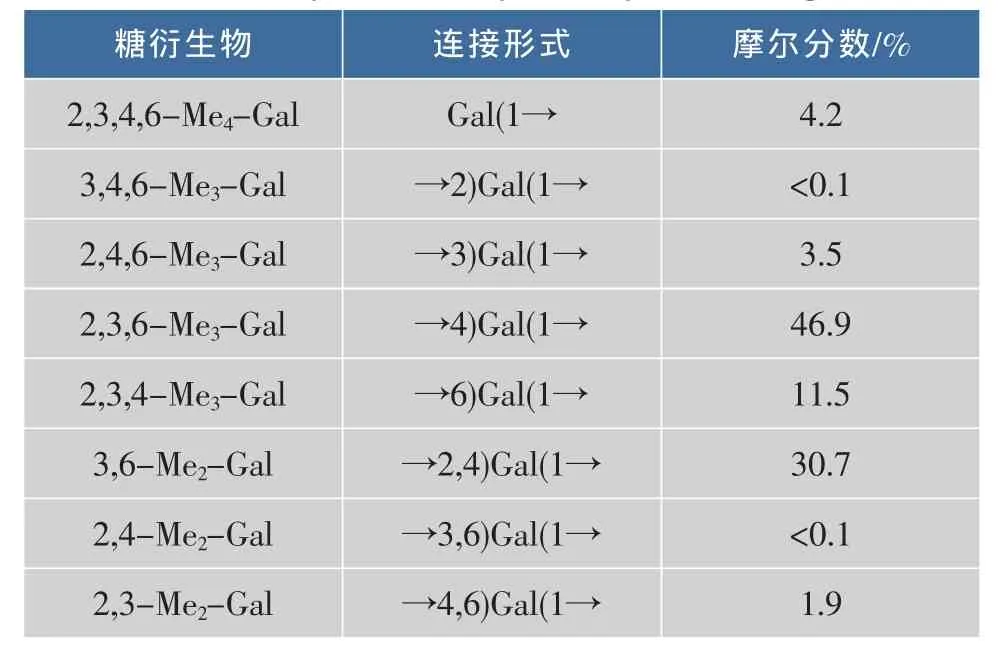
表1 半乳聚糖的甲基化分析Table 1 Methylation analysis of synthesized galactan
2.5 核磁共振波譜分析
核磁共振波譜是多糖結構解析過程中的一個強有力的工具,能夠提供更多糖殘基的平面結構和立體構型信息。在半乳聚糖的1H-NMR譜(圖4(a))中,大多數的端基質子化學位移出現在δ 5.02~5.30(J=0~2 Hz)處,而少數端基質子信號位于δ 4.49(d,J=5.5 Hz)和δ 4.63(d,J=6.0 Hz),δ 3.5~4.4為半乳糖殘基H-2~H-6的質子信號。一般地,在以重水為溶劑的核磁測試中,吡喃型糖殘基的α-構型端基質子出現在δ 5~6,偶合常數為2~4 Hz;而β-構型端基質子出現在δ 4~5,偶合常數為6~8 Hz[18]。因此,合成半乳聚糖主要以α-構型為主,同時有少量的β-構型半乳糖殘基。這與王海松等[9]的分析結果截然相反,可能是由于不同氘代試劑對質子化學位移的影響,從而導致分析結果不一致。在王海松等合成的半乳聚糖的1H NMR圖譜[9]分析中,采用氘代二甲基亞砜為溶劑,致使端基質子的核磁信號集體往高場移動約0.3×10-6,卻以重水為測試試劑時的構型判別規律為參照,這是不恰當的。在13CNMR譜中,半乳聚糖的端基碳化學位移主要集中在δ 99~110,而C-2~C-6的信號集中在δ63~85,未觀察到羰基或羧基的化學位移信號(圖4(b))。
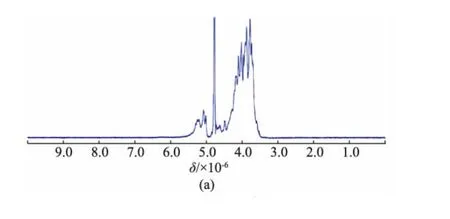
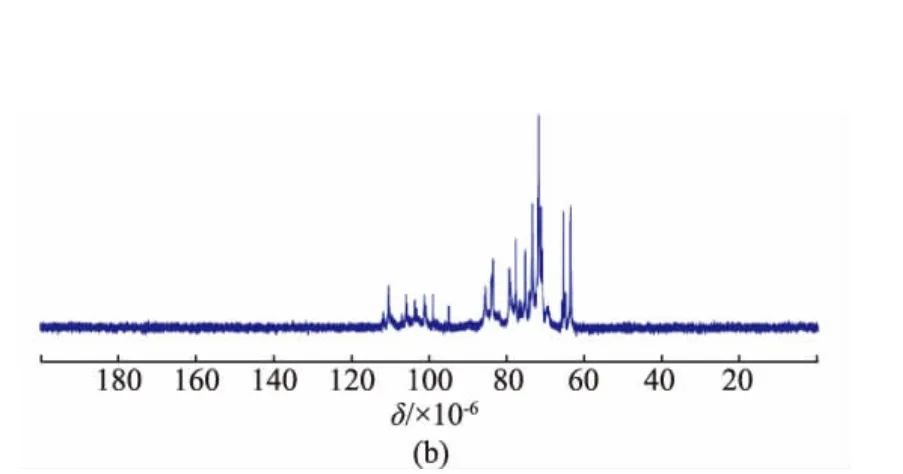
圖4 聚半乳糖的1H(500 MHz)和13C(125 MHz)核磁共振譜圖Fig.41H and13C NMR spectra of synthesized galactan(500 MHz for1H NMR and 125 MHz for13C NMR in D2O)

圖5 半乳聚糖的1H-1H COSY和TOCSY核磁共振譜圖Fig.51H-1H COSY and TOCSY spectra of synthesized galactan
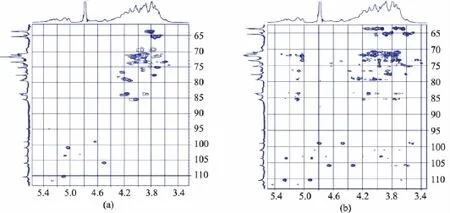
圖6 半乳聚糖的HSQC和HMBC核磁共振譜圖Fig.6 HSQC and HMBC spectra of synthesized galactan
根據半乳糖殘基H-1/H-2/H-3/H-4/H-5/H2-6質子的相關性,分析反映鄰近質子偶合相關性的1H-1H COSY譜和反映同一自旋體系中質子標量偶合相關性的TOCSY譜(圖5),對不同連接型半乳糖殘基的質子信號進行歸屬,結果如表2所示。以β-構型半乳糖殘基端基質子信號δ 4.49為例,在TOCSY譜中,δ 4.49與δ 3.60,3.70,3.98,4.02有較強的相關信號,表明這5個質子存在于同一個半乳糖殘基中;結合1H-1H COSY譜中δ 4.49與3.60的相關信號,推斷H-2的化學位移為δ 3.60,進而根據H-2/H-3的相關信號,推斷H-3的化學位移為δ 3.70;類似地,最終推導了整個半乳糖殘基的質子信號。根據HSQC譜中氫、碳核磁信號的相關性(圖6(a)),歸屬了不同質子信號對應的碳化學位移(表2),并根據HMBC譜中(圖6(b)),氫、碳信號之間的長程偶合(3J)相關,對歸屬的質子信號進行驗證,并推斷半乳糖殘基中的取代位點。端基質子δ 5.02與δ 73.5(C-6)有強HMBC相關信號,表明半乳聚糖中存在6位羥基取代的糖殘基,而往低場移動了約8×10-6的C-6铘化學位移,進一步表明結構中存在→6)Gal(1→結構片段。
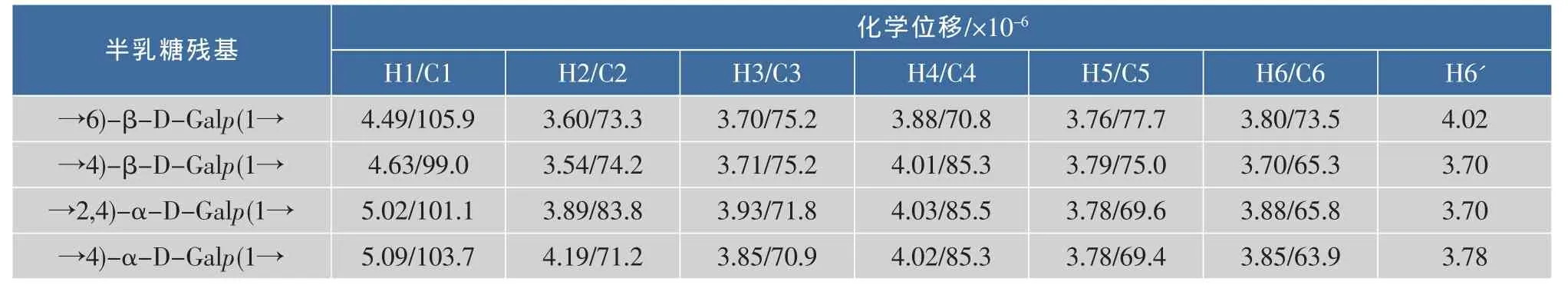
表2 半乳聚糖主要糖殘基的化學位移數據Table 2 Chemical shifts of major galactosyl residues in synthesized galactan
對一維 (1H,13C NMR)和二維 (1H-1H COSY,TOCSY,HSQC和HMBC)核磁圖譜的綜合分析,最終解析了→6)-β-D-Galp(1→,→4)-β-D-Galp(1→,→4)-α-D-Galp(1→和→2,4)-α-D-Galp(1→糖殘基信號(表2),對照半乳聚糖的相對分子質量和甲基化分析結果(表1),表明微波輔助、磷酸催化合成的半乳聚糖結構中存在較多的→4)-DGalp(1→和→2,4)-α-D-Galp(1→片段,推測該糖鏈為多分支的鏈狀結構。
3 結語
在微波輻照條件下,采用磷酸代替雜多酸能夠快速、高效地催化D-半乳糖的縮合,純化后半乳聚糖的得率為90.4%。反應生成的半乳聚糖為分支鏈狀結構,平均聚合度為16,立體構型以α為主,新糖苷鍵的生成主要發生在4位、2位和6位的羥基上。通過對半乳聚糖的結構分析表明,微波輔助、磷酸催化半乳糖縮合過程中,不同位置的羥基反應活性不同,若能對目標羥基進行保護性修飾,則有可能進一步提高反應的選擇性。微波輔助合成半乳聚糖結構的闡明,也為進一步研究和理解其生物活性提供了基礎。
[1]CEDRIC D,TARATRA Andree F,PHILIPPE M.Galactans:an overview of their most important sourcing and applications as natural polysaccharides[J].Brazilian Archives of Biology and Technology,2011,54(6):1075-1092.
[2]CARLUCCI M J,SCOLARO L A,ERREA M I,et al.Antiviral activity of natural sulphated galactans on herpes virus multiplication in cell culture[J].Planta Medicine,1997,63(5):429-432.
[3]CHATTOPADHYAY K,MATEU Cecilia G,MANDAL P,et al.Galactan sulfate of Grateloupia indica:isolation,structural features and antiviral activity[J].Phytochemistry,2007,68(10):1428-1435.
[4]FARIAS W R,VALENTE A P,PEREIRA M S,et al.Structure and anticoagulant activity of sulphated galactans:isolation of a unique sulphated galactan from the red algae Botryocladia occidentalis and comparison of its anticoagulant action with that of sulphated galactans from invertebrates[J].Journal of Bological Chemistry,2000,275(38):299-307.
[5]COOMBE D R,PARISH C R,RAMSHAW I A,et al.Analysis of the inhibition of tumour metastasis by sulphated polysaccharides[J].International Journal of Cancer,1987,39(1):82-88.
[6]WU Chengfei,LI Yan,LE Guowei,et al.Effect of the composing and monosaccharides ratio changing of oligosaccharides on free radical scavenging activity and Lactobacillus proliferation[J].Science and Technology of Food Industry,2014,35:49-56.(in Chinese)
[7]WANG H,SHI Y,LE G.Rapid microwave-assisted synthesis of polydextrose and identification of structure and function[J]. Carbohydrate Polymers,2014,113:225-230.
[8]WANG H,CHENG X,SHI Y,et al.Preparation and structural characterization of poly-mannose synthesized by phosphoric acid catalyzation under microwave irradiation[J].Carbohydrate Polymers,2015,121:355-361.
[9]WANG Haisong,SHI Yonghui,LE Guowei.Microwave-assisted synthesis and structural analysis of galactan[J].Food Science,2014,35:35-39.(in Chinese)
[10]WONG C H.Protein glycosylation:new challenges and opportunities[J].Journal of Organic Chemistry,2005,70:4219-4225.
[11]HSU C H,HUNG S C,WU C Y,et al.Toward automated oligosaccharide synthesis[J].Angewandte Chemie,International Edition,2011,50:11872-11923.
[12]CHENG Xiangrong,WANG Wei,SHI Yonghui,et al.Microwave assisted synthesis of mannose oligosaccharide intermediates[J]. Journal of Food Science and Biotechnology,2015,34(1):21-27.(in Chinese)
[13]GUO Jinlong,CHEN Youjun,SUN Guoqin,et al.Study on phenol-sulfuric acid method for determination of polysaccharide content in Pleurotus eryngii[J].Food Science,2008,29:555-558.(in Chinese)
[14]HAKOMORI S.Rapid permethylation of glycolipids and polysaccharides,catalyzed by methylsulfinyl carbanion in dimethyl sulfoxide[J].Journal of Biochemistry,1964,55(2):205-208.
[15]HU P,XUE R,LI Z,et al.Structural investigation and immunological activity of a heteropolysaccharide from Sargassum fusiforme [J].Carbohydrate Research,2014,390:28-32.
[16]ZHANG Q,LI N,LIU X,et al.The structure of a sulfated galactan from Porphyra haitanensis and its in vivo antioxidant activity [J].Carbohydrate Research,2004,339:105-111.
[17]PEREIRA M G,BENEVIDES N M B,MELO M R S,et al.Structure and anticoagulant activity of a sulfated galactan from the red alga,Gelidium crinale.Is there a specific structural requirement for the anticoagulant action?[J].Carbohydrate Research,2005,340:2015-2023.
[18]CUI S W.Food carbohydrates chemistry,physical properties,and applications[M].CRC Press:Taylor&Francis Group,2005:154-155.
會議消息
Preparation and Structural Characterization of Galactans Synthesized by Phosphoric Acid Catalyzation under Microwave Irradiation
JI Gao1, WANG Haisong1, DING Yinyi1, CHENG Xiangrong*1,2, LE Guowei1,2
(1.School of Food Science and Technology,Jiangnan University,Wuxi 214122,China;2.State Key Laboratory of Food Science and Technology,Jiangnan University,Wuxi 214122,China)
Galactans synthesized by phosphoric acid catalysis and microwave irradiation were studied for their chemical structures and regio-and stereo-selectivities during the polymerization.A series of analyses including high-performance gel-permeation chromatography,infrared spectroscopy,methylation analysis and NMR spectroscopies(1H,13C,1H-1H COSY,TOCSY,HSQC,HMBC)were used.Results showed that the degree of galactosyl polymerization was 16 and the -galactopyranosyl residues were dominant mainly with→4)-α-D-Galp(1→and→2,4)-α-D-Galp(1→fragments.Structural analyses further indicated that the activities of hydroxyls in galactose were different and the 4-OH exhibited the highest potency.
microwave,phosphoric acid,galactan,structural analyses
TS 245.9
A
1673—1689(2017)01—0027—07
2015-03-24
國家“十二五”科技支撐計劃項目(2012BAD33B05)。
*通信作者:成向榮(1985—),男,浙江永康人,醫學博士,副教授,主要從事食品營養與功能因子研究。E-mail:cheng-xiangrong@hotmail.com
紀 高,王海松,丁寅翼,等.微波輔助、磷酸催化合成半乳聚糖的結構分析[J].食品與生物技術學報,2017,36(01):27-33.
猜你喜歡
鴨綠江(2021年35期)2021-04-19 12:24:18
考試與評價·高一版(2020年6期)2020-11-02 02:45:24
媽媽寶寶(2019年10期)2019-10-26 02:45:34
中國生殖健康(2019年3期)2019-02-01 06:12:26
鐵道通信信號(2018年11期)2019-01-19 01:15:08
電子制作(2018年11期)2018-08-04 03:25:42
鐵道通信信號(2018年2期)2018-04-18 12:18:10
鐵道通信信號(2016年11期)2016-06-01 12:11:32
鑿巖機械氣動工具(2016年3期)2016-03-01 04:00:25
中國病理生理雜志(2015年8期)2015-12-21 12:38:06
