線粒體基因T13094C突變致伴頸髓病變的線粒體腦肌病伴高乳酸血癥和卒中樣發作/Leigh疊加綜合征的臨床、影像、病理及基因突變特點(附1例報告)
2017-04-27 02:36:46劉改玲張允李園園都愛蓮
臨床神經病學雜志 2017年2期
劉改玲,張允,李園園,都愛蓮
線粒體基因T13094C突變致伴頸髓病變的線粒體腦肌病伴高乳酸血癥和卒中樣發作/Leigh疊加綜合征的臨床、影像、病理及基因突變特點(附1例報告)
劉改玲,張允,李園園,都愛蓮
目的 探討線粒體基因T13094C突變致伴有頸髓病變的線粒體腦肌病伴高乳酸血癥和卒中樣發作(MELAS)/Leigh疊加綜合征的臨床、影像、病理及基因突變特點。方法 回顧性分析1例伴頸髓病變的MELAS/Leigh疊加綜合征患者的臨床資料。結果 患者為14歲女性,以雙下肢麻木、抽搐起病,癥狀反復發作并進展,逐漸出現嘔吐、眼球運動障礙、共濟失調、乳酸酸中毒、易激惹、呼吸衰竭、昏迷等,于發病后20個月時死于癲癇持續狀態。影像學表現為雙側大腦半球、C2-6段頸髓、丘腦、雙側小腦半球、中腦、腦橋、延髓等處不斷變化的新病灶。肌肉病理學檢查示Gomori染色可見破碎紅纖維,琥珀酸脫氫酶染色可見周邊深染的肌纖維,電鏡示明顯的線粒體數量和形態異常。血和肌肉線粒體基因測序示T13094C雜合突變。結論 線粒體基因T13094G突變致MELAS/Leigh綜合征出現頸髓病變為線粒體病所罕見;持續進展乃至死亡的病程不同于A3243G突變患者。
線粒體腦肌病伴高乳酸血癥和卒中樣發作;Leigh綜合征;疊加綜合征;頸髓病變;T13094C基因突變
線粒體腦肌病是一種異質性極高的遺傳代謝性疾病,臨床表現復雜多樣,與其他神經系統疾病如病毒性腦炎、多發性硬化、脊髓小腦性共濟失調等很難區分。線粒體病的不同亞型之間也有相互重疊,最多見的疊加綜合征是線粒體腦肌病伴高乳酸血癥和卒中樣發作(MELAS)/Leigh疊加綜合征。導致疊加綜合征的致病基因很多,如線粒體基因G13513A[1]或A13084T[2]等。本研究回顧性分析1例由線粒體基因T13094C突變導致的伴有頸髓病變的MEALS/Leigh疊加綜合征,并探討其臨床、影像、病理及基因突變特點。
1 臨床資料
患者,女,14歲,祖籍浙江。因“反復肢體麻木、抽搐11個月,伴頭痛、嘔吐3個月”于2013年2月22日入院。足月順產,自幼運動能力差,100米短跑不及格。患者于2012年3月22日爬山后出現雙下肢麻木、緊繃感,由肢體遠端向近端發展,左下肢體抽搐伴意識障礙發作1次,走路不穩,無發熱。在當地醫院查頭頸部MRI示右側大腦腳、額葉皮質見片狀稍長T1、長T2信號影,C2-6椎體平面頸髓條狀等T1、稍長T2信號影(圖1)。腰椎穿刺查CSF細胞數1×106個/L,葡萄糖2.8 mmol/L,氯118 mmol/L,蛋白314 mg/L。視頻EEG示兩側前額、前顳、枕區4~5 Hz慢/慢波陣發性出現。考慮“播散性腦脊髓炎”,予甲潑尼龍沖擊、丙種球蛋白治療,癥狀好轉出院。2012年12月3日,患者晚自習后出現惡心嘔吐,四肢抽搐、雙眼凝視、小便失禁,右側肢體麻木無力。于當地醫院行頭部MRI示右側大腦腳、右側額葉病灶增大,左側小腦半球、雙側丘腦、雙側額葉皮質見新增病灶,考慮“多發性硬化”,予甲潑尼龍沖擊治療,并拉莫三嗪抗癲癇治療,癥狀緩解出院。2013年2月22日,患者再次出右側肢體無力,伴右側肢體和面部抽搐,從右手開始,持續數秒至2 min不等,無意識障礙。遂至我院就診,查CSF常規、生化、IgG指數均正常,CSF乳酸2.8 mmol/L;CSF送至華山醫院查寡克隆帶、水通道蛋白4均陰性;當天同步查血乳酸2.1 mmol/L。查體:顱神經陰性,右側肢體肌力Ⅳ級,右側肢體觸覺減退,雙側Babinski征陰性。入院后查頭顱MRI示雙側頂葉新近異常信號。肌酸激酶正常。EMG未見明顯異常。骨骼肌肌肉病理檢查示Gomori染色可見破碎紅纖維,琥珀酸脫氫酶染色可見周邊深染的肌纖維,電鏡下可見大量的線粒體在肌漿膜下聚集,線粒體腫脹、內部結構紊亂或消失(圖2)。基因檢測示血液中DNA存在低比例的T13094C異質性突變,但在患者肌肉組織DNA中檢測到高比例T13094C突變(圖3)。確診為:MEALS/Leigh疊加綜合征。給予輔酶Q10、ATP、左卡尼汀等治療后癥狀好轉,于2013年3月15日出院。對患者的母親、姨媽、外婆和胞弟4位母系成員線粒體基因13094位點進行Sanger測序,均未發現類似突變。2013年4月13日患者抽搐伴意識不清、呼吸急促再次來我院急診,急查血常規WBC 41.4×109,血氣分析示pH 6.848,二氧化碳分壓57.0 mmHg(1 mmHg=0.133 kPa),氧分壓65.5 mmHg, 血乳酸18 mmol/L,血糖15.9 mmol/L,予緊急氣管插管,碳酸氫鈉、抗感染等治療4 h后好轉并拔管。隨后收住入院,查頭顱MRI示左側額葉、雙側丘腦和中腦、右側小腦半球、左側延髓異常信號(圖4)。給予輔酶Q10、左卡尼汀口服液治療。4月18日患者出現行走不穩,復查頭顱MRI示雙側中腦、小腦、頂葉多發新近病灶,予加用L-精氨酸針劑。4月22日患者出現頭頸部和四肢肌陣攣,并出現興奮、言語增多,易激惹,雙眼下視障礙、外展困難,共濟失調,雙眼水平眼震。在原有藥物基礎上加用ATP片等治療后部分好轉,于4月30日出院。2013年7月28日患者再次出現頭暈、頭痛,伴惡心,無暈厥及抽搐,當地醫院監測血壓80~90 mmHg/40~60 mmHg,予多巴胺針靜脈推注升壓治療后轉入我院。7月29日入院查體:神志清,精神萎,反應較遲鈍,雙眼垂直運動障礙,水平眼震,肢體肌力Ⅴ-級。7月30日頭顱MRI示雙側中腦、丘腦、小腦、延髓、頂葉多發病灶。8月5日患者出現雙眼瞼下垂,雙眼球向上、向下、內收、外展均活動受限。8月6日患者訴呼吸困難、頭暈乏力。8月7日患者出現嗜睡、反應遲鈍、口齒含糊,疼痛刺激時有呻吟,雙側下肢疼痛刺激有逃避,四肢末端冰冷,心電監護示心率150次/min,血氧飽和度為99%,呼吸27次/min,血壓140 mmHg /83 mmHg。給予補液、糾正酸中毒、抗感染、控制心室率及營養支持治療,8月8日患者持續昏睡狀態,GCS評分為7分,雙側瞳孔對光反應遲鈍。8月11日凌晨5:08時突發呼吸微弱,血壓下降,神志不清。查體:體溫39.0 ℃,血壓88 mmHg/44 mmHg,血氧飽和度60%,心率160次/min,血氣分析示pH 6.984、二氧化碳分壓114.0 mmHg。予緊急氣管插管,呼吸機輔助呼吸,抗感染、糾正酸中毒等治療。8月20日查體:淺昏迷,自主呼吸存在,偶見肢體自發躁動,下肢疼痛刺激可回縮,雙側瞳孔直徑2 mm,對光反射消失,雙側Babinski征陽性。9月20日病情恢復到呼之能睜眼、有情感反應、右手能拉扯衣服,予脫呼吸機出院回家。2013年10月28日患者再次發作四肢抽搐,因癲癇持續狀態在當地醫院死亡。

圖1 患者2012年3月22日頭頸部MRI檢查示頸髓腹側長條狀異常信號,雙側丘腦、雙側中腦異常信號

圖2 患者骨骼肌病理檢查。A:Gomori染色看到破碎紅纖維;B:琥珀酸脫氫酶染色見到周邊深染的肌纖維;C:電鏡下可見大量的線粒體在肌漿膜下聚集,線粒體腫脹、內部結構紊亂或消失
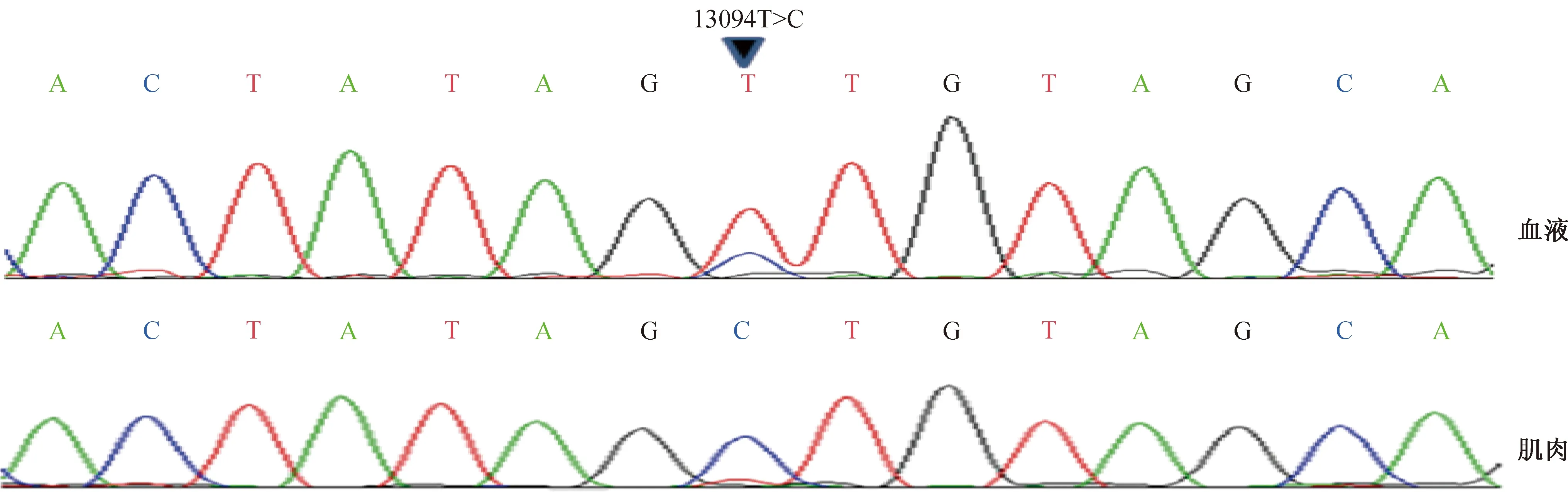
圖3 患者基因檢測示外周血檢測出線粒體基因T13094C 異質性突變,肌肉中T13094C突變的比率接近100%

圖4 患者2013年4月13日頭部MRI檢查示左側額葉、雙側丘腦和中腦、右側小腦半球、左側延髓異常信號
2 討 論
本例患者早期發育正常,14歲開始以反復發作的頭痛嘔吐、抽搐、肢體麻木無力起病,同時出現了丘腦、中腦和頸髓長節段的異常信號,早期糖皮質激素治療后好轉,臨床表現極易誤診為多發性硬化。后期反復發作時出現了眼肌麻痹、眼球震顫、共濟失調、乳酸酸中毒伴呼吸衰竭等典型表現,影像學表現以丘腦、中腦、延髓和小腦為主,有早期的脊髓受累,并伴有片狀的額頂葉皮質病灶,同時具有MELAS綜合征和Leigh綜合征的影像學特點;單純的Leigh綜合征破碎紅纖維陽性率較低,而本患者病理表現見到較多的典型的破碎紅纖維也支持MELAS綜合征的診斷,但起病后迅速進展、乃至死亡更符合Leigh綜合征的臨床特點,故診斷為MELAS/Leigh疊加綜合征。外周血和肌肉DNA基因檢測明確了線粒體ND5基因T13094C突變,導致編碼NADH 脫氫酶 (復合體Ⅰ)的 253位點上的纈氨酸變為丙氨酸(p.V253A)。關于線粒體基因T13094C突變作為致病突變于2009年由Valente等[3]首次報道過,主要臨床表型為Leigh綜合征[4]、小腦性共濟失調、進行性眼外肌麻痹[3,5]或者MELAS/Leigh疊加綜合征[6]。這個位點的突變血液中檢出率低,很少在家系成員中檢出陽性結果,本例患者血液中突變比例很低,但在肌肉中突變比例卻接近100%,患者的母親、外婆、姨媽和弟弟血液中均未檢測到突變,這與國際報道[3]的線粒體基因T13094C突變特點相吻合。該基因位點突變致病的另一突出特點是進展迅速,本例患者發病后的19個月內共發作10余次,發作間隙越來越短,在應用輔酶Q10、左卡尼汀、ATP等線粒體腦肌病治療藥物干預后仍持續進展,符合Leigh綜合征的病程特點。國外報道[3-4]的幾例Leigh綜合征、小腦性共濟失調、進行性眼外肌麻痹和MELAS/Leigh疊加綜合征也表現出了類似的進展過程。這種病程特點的發生機制尚待進一步研究。
[1]Shanske S, Coku J, Lu J, et al. The G13513A mutation in the ND5 gene of mitochondrial DNA as a common cause of MELAS or Leigh syndrome: evidence from 12 cases [J]. Arch Neurol, 2008, 65: 368.
[2]Crimi M,Galbiatis,Moroni I, et al.A missense mutation in the mitoehondrial ND5 gene associated with a Leigh/MELAS overlap syndrome [J].Neurology, 2003, 60: 1857.
[3]Valente L, Piga D, Lamantea E, et al. Identification of novel mutations in five patients with mitochondrial encephalomyopathy [J]. Biochim Biophys Acta, 2009, 1787: 491.
[4]Swalwell H, Kirby DM, Blakely EL, et al. Respiratory chain complex Ⅰ deficiency caused by mitochondrial DNA mutations [J]. European Journal of Human Genetics, 2011, 19: 769.
[5]Calvo SE, Tucker EJ, Compton AG, et al. High-throughput, pooled sequencing identifies mutations in NUBPL and FOXRED1 in human complex Ⅰ deficiency [J]. Nat Genet, 2010, 42: 851.
[6]Lax NZ, Hepplewhite PD, Reeve AK, et al. Cerebellar ataxia in patients with mitochondrial DNA disease: a molecular clinicopathological study [J]. J Neuropathol Exp Neurol, 2012, 71: 148.
Clinical, imaging, pathological and gene mutation features of mitochondrial gene T13094C mutation caused mitochondrial encephalomyopathy with lactic acidosis and stroke like episodes/Leigh overlap syndrome with cervical spinal lesion (report of 1 case)
LIUGai-ling,ZHANGYun,LIYuan-yuan,etal.
DepartmentofNeurology,TongrenHospital,ShanghaiJiaoTongUniversitySchoolofMedcine,Shanghai200336,China
Objective To observe the clinical, imaging, pathological and gene mutation features of mitochondrial gene T13094C mutation caused mitochondrial encephalomyopathy with lactic acidosis and stroke like episodes (MELAS)/Leigh overlap syndrome with cervical spinal lesion.Methods The clinical data of one MELAS/Leigh overlap syndrome patient with cervical spinal lesion was analyzed retrospectively.Results The 14-years-old female patient first presented with numbness and convulsion of both legs. The symptoms were recurrent and progress, and then vomiting, ophthalmoplegia, ataxia, lactate acidosis, irritability, respiratory failure and coma were gradually appeared. At 20 months after onset, she died of status epileptic. The imaging performances were changing of new lesions at bilateral cerebral hemisphere, C2-6section of the cervical spinal, thalamus, bilateral cerebellum, midbrain, pons, medulla oblongata. The main muscle pathological findings were red rag fibers on Gomri staining, enhanced stained fibers on succinodehydrogenase staining, and dramatic mitochondrion abnormality on electron microscopy. Mitochondrial gene sequencing in blood and muscle showed T13094C heterozygous mutation.Conclusions Mitochondrial gene T13094C mutation caused MELAS/Leigh superposition syndrome showed cervical spinal lesion is rare in mitochondrial disease. The disease course continuous progress to die is different from A3243G gene mutation patients.
mitochondrial encephalomyopathy with lactic acidosis and stroke like episodes; Leigh syndrome; superposition syndrome; cervical spinal lesion; T13094C gene mutations
國家自然科學基金(81200967);上海市衛生和計劃生育委員會基金(201540050)
200336上海交通大學醫學院附屬同仁醫院神經內科[劉改玲,張允,李園園,都愛蓮(原在浙江大學醫學院附屬第二醫院神經內科)]
張允,都愛蓮
R746
A
1004-1648(2017)02-0089-04
2016-06-01
2016-06-24)
