臭常山理化特征及其脂溶性生物堿的提取工藝
2017-05-17 17:37:48呂慶銀趙偉李岑李雄張振
江蘇農業科學 2016年1期
呂慶銀 趙偉 李岑 李雄 張振

摘要:先對臭常山的水分、總灰分、醇浸出物及其TLC、HPLC指紋圖譜進行研究,明晰試驗藥料的理化特征,然后采用超聲波輔助乙醇提取法對臭常山脂溶性生物堿的提取工藝進行正交優化。結果表明,本試驗材料臭常山的水分、醇溶性浸出物、總灰分含量分別為8.21%、6.22%、6.20%;在單因素的基礎上,正交試驗確定的最佳提取工藝條件為臭常山粗粉400 g,料液比1 g:20 mL,65%乙醇超聲提取80 min/次,超聲提取3次,脂溶性生物堿的提取率為0.385%。
關鍵詞:臭常山;指紋圖譜;脂溶性生物堿;正交試驗
中圖分類號:R284.2 文獻標志碼:A 文章編號:1002—1302(2016)01—0279—04
蕓香科(Rutaceae)臭常山屬臭常山(Orixa japonica Thunb.),別稱日本常山、臭山羊,其主要化學成分為香豆素和喹啉類生物堿。目前關于其研究主要集中在所含化學成分方面,現從臭常山的根、莖、葉等部位分離到的已知喹啉類生物堿有:常山堿、香草木堿、茵芋堿、月蕓香酮堿、香草木堿、去甲和常山堿、加錫彌羅果堿、花椒毒素等。試驗前期研究發現,這些化學成分中的一些喹啉類脂溶性生物堿具有抗肺炎克雷伯氏桿菌及金黃色葡萄球菌等抑菌活性。作為貴州民間中藥材,常用來治療風熱感冒、咳嗽、風濕關節痛等病癥,其理化特征和提取工藝方面的研究未見國內外文獻報道。為富集其有效成分,在對試驗臭常山理化特征進行研究后,進行有效成分的提取分離工藝研究,以期明確試驗材料的理化特征,并獲得脂溶性生物堿的最佳提取工藝途徑,為推進臭常山中藥現代化研究提供一定的理論基礎。
1試驗材料
1.1材料與試劑
新鮮植物整株經貴州大學茍光前教授鑒定為蕓香科植物臭常山(Orixajaponica Thunb.);色譜乙腈及色譜甲醇(美國天地公司)、超純水(浙江杭州哇哈哈集團有限公司);蒸餾水;其余試劑均為分析純。
1.2儀器和設備
UltiMaterTM3000 DGLC雙三元液相色譜儀,富集柱(Ther-mo Fisher HyperSep Retain-CX,20 mm×3.0 mm),DAD檢測器,分析柱(Acclaim 120 PAⅡ,C18,5μ×m,4.6 mm×250 mm),以上均為美國Thermo Fisher Scientific公司產品;變色龍7.2工作站;SPJX-4-10型高溫箱式電阻爐(上海錦昱科學儀器有限公司);WT5001電子天平(江蘇常州萬泰天平儀器有限公司);ES-E210B電子分析天平(河南鄭州南北儀器設備有限公司);潔康PS-100A超聲波清洗機(廣東東莞市潔康超聲波設備有限公司);SHZ-D(Ⅲ)型循環水式真空泵、RE-501升降恒溫水浴鍋、RE-501型旋轉蒸發器及DLSB-10/20℃型低溫冷卻循環泵(均為上海鷹迪儀器設備有限公司);GF254硅膠板(山東青島海洋化工廠),ZF-2型三用紫外儀(上海市安亭電子儀器廠)。
2試驗方法
2.1材料預處理
將新鮮的臭常山整株截短、洗凈,然后晾干、粉碎,自然風干后備用。
2.2臭常山水分、總灰分、醇浸出物的測定
水分含量測定采用中國藥典一部附錄ⅨH水分測定法項下的烘干法;醇浸出物測定采用藥典一部附錄Ⅹ A項下醇提取物熱浸法;灰分測定采用藥典附錄ⅨK項下的總灰分測定法。
2.3 TLC指紋圖譜
準確稱取臭常山全株粗粉100 g,以1 g:5 mL的料液比加入70%乙醇,超聲提取30 min,過濾,濃縮,吸取少許濃縮液點于硅膠GF254板,以石油醚:丙酮=8:2為展開劑,展開,晾干,于紫外燈(257.5 am)下檢視。
2.4 HPLC指紋圖譜
2.4.1測試樣品制備 準確稱取過二號篩的臭常山粉末2.408 9 g,加70%乙醇60 mL置250 mL錐形瓶中,密塞稱定質量,靜置1 h,然后連接回流冷凝管,并保持微沸1 h,放冷后,取下錐形瓶,密塞,再稱定質量,用70%乙醇補足減失的質量,搖勻,用干燥過濾器過濾,取濾液過0.45μm針頭過濾器,即得測試樣品。
2.4.2對照品制備分別精確稱取月蕓香酮堿、香草木堿(實驗室自制,純度>95%),加入甲醇后制成100μg/mL的對照品溶液。
2.4.3色譜條件色譜柱為C18(250 mm×4.6 mm,5μm),流動相為乙腈-水(體積比1:1),體積流量1.00 mL/min,進樣量20μL,柱溫30℃,檢測波長266 nm。
2.5脂溶性生物堿的制備及定量測定
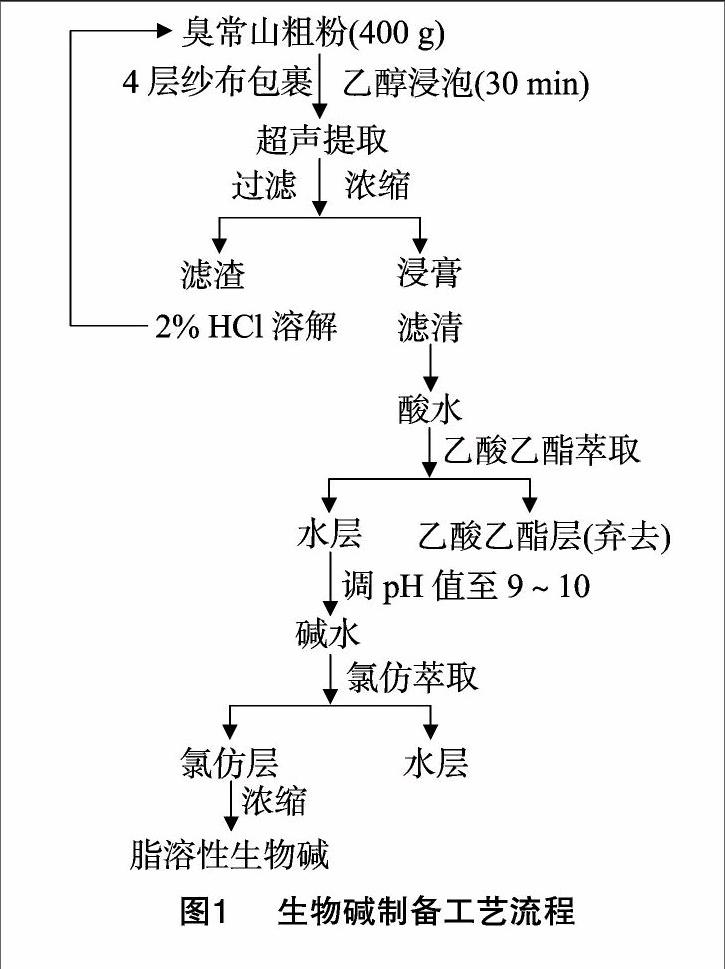
(1)醇粗提物提取。準確稱取臭常山粗粉400 g,4層紗布包裹,按試驗所需的濃度和料液比加入乙醇進行超聲提取,然后過濾,濾渣進行重復提取,濾液濃縮成浸膏,干燥,即得粗提物。(2)脂溶性生物堿的制備和測定。取適量的2%HCl溶解上述浸膏,濾清酸水,等體積的乙酸乙酯萃取酸水3次,棄去乙酸乙酯層,25%氨水調節pH值為9~10,等體積氯仿萃取3次,合并氯仿層,濃縮,回收氯仿,殘留物即為脂溶性生物堿,干燥,稱質量。
2.6脂溶性生物堿的定性檢測
生物堿的定性鑒定方法有很多,本試驗采用碘化鉍鉀顯色法和薄層色譜法,具體方法如下:(1)取“2.5”節下制得的脂溶性生物堿3~5 mg,適量氯仿溶解;(2)毛細吸管吸取上述溶液點于硅膠GF254板上;(3)以石油醚-丙酮(8:2)為展開劑展開,取出,晾干,紫外燈(257.5 nm)下檢視;(4)噴碘化鉍鉀顯色液,晾干,日光下觀察;日光下有多個橘黃色或橙色斑點,紫外燈下有多個熒光亮點,即可判斷酸堿處理結合溶劑萃取后得到的是脂溶性生物堿。
3結果與分析
3.1試驗用臭常山的水分、總灰分、醇浸出物含量比較
由表1可知,本試驗用臭常山的7次測試樣品中,最高水分含量和最低水分含量相差0.07百分點,兩者相差不到0.1百分點,表明水分含量測定方法重復性良好;總灰分含量最高的為6.30%,最低的為6.11%,兩者相差0.19%,這可能是每次取樣量的多少不同,導致需要高溫熾灼的次數不同,而測試的精確度只需到0.01g,所以操作誤差相對會變大,試驗重復性差些;70%醇浸出物含量最高的為6.27%,最低的為6.11%,兩者相差不到0.2百分點,醇浸出物含量測定穩定性和重復性良好。
3.2指紋圖譜
3.2.1 TLC指紋圖譜 按上述“2.3”節的條件,毛細管分別吸取3次樣品溶液少許,點于同一塊GF254板上,對試驗材料進行TLC指紋圖譜分析,結果(圖2)表明,同一樣點在以石油醚-丙酮(8:2)為展開劑,經展開后得到了多個熒光點,表明此展開體系適用于本樣品的TLC指紋圖譜構建;且3次點樣得到的顯熒光點位置幾乎一致,表明試驗重復性良好。
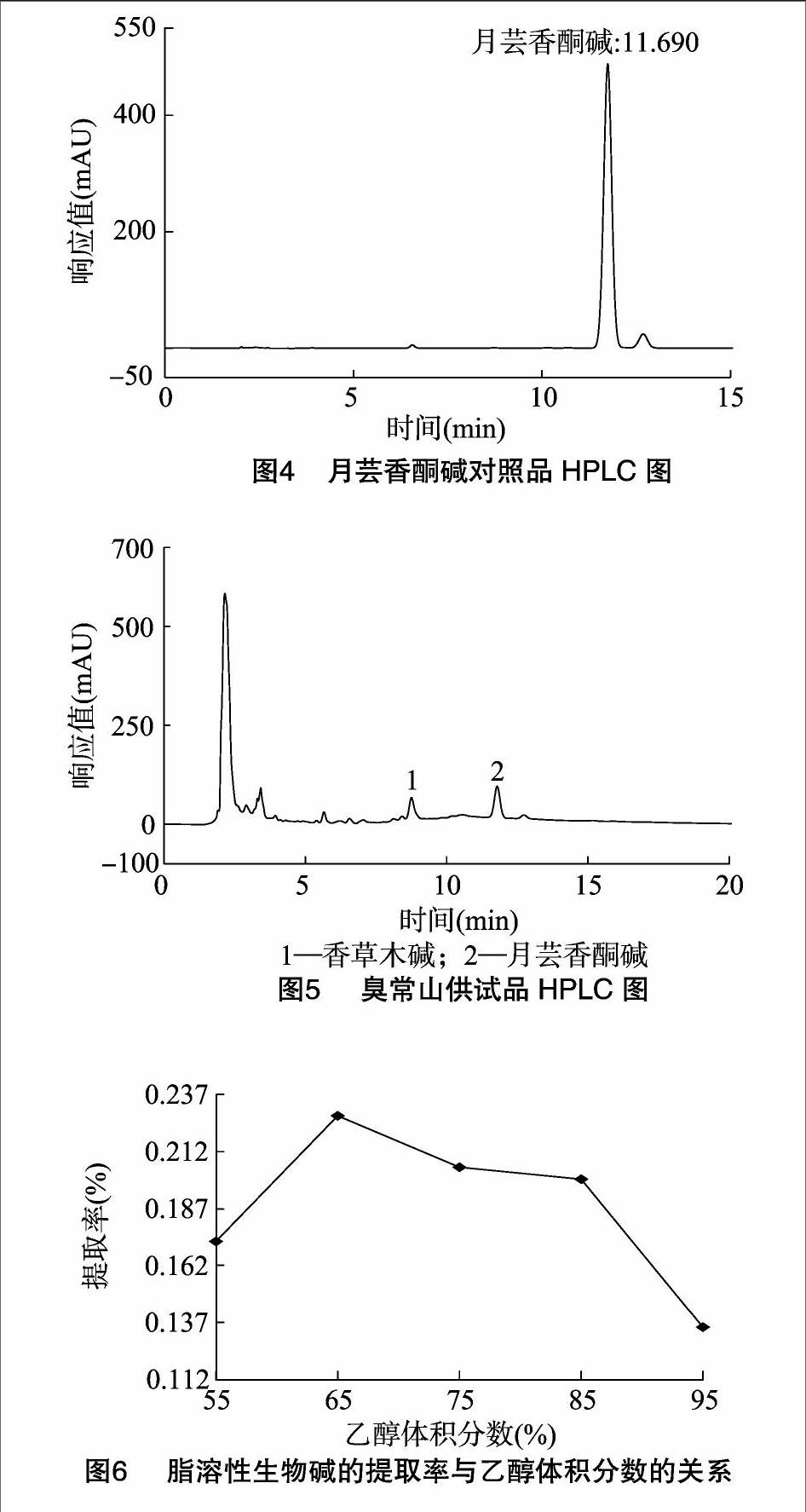
3.2.2 HPLC指紋圖譜 按上述條件,對試驗材料進行HPLC指紋圖譜分析。分別吸取對照品和供試品溶液各20μL,分別注入高效液相色譜儀,記錄色譜圖,結果見圖3至圖5。對照品香草木堿和月蕓香酮堿的保留時間分別為8 min左右和11.5 min左右,峰分離度好,峰形對稱。
3.2脂溶性生物堿提取的單因素試驗
3.2.1乙醇濃度對脂溶性生物堿提取的影響 準確稱取臭常山粗粉400 g 5份,以料液比為1 g:10 mL,分別加入體積分數為55%、65%、75%、85%、95%的乙醇,超聲提取30 min,按“2.5”節下的方法對各組脂溶性生物堿進行定性和定量測定,考察不同濃度的乙醇對脂溶性生物堿提取率的影響,試驗結果見圖6。
由圖6可知,脂溶性生物堿的提取率在乙醇體積分數為65%時達到最大,當乙醇的體積分數超過65%時,隨著乙醇體積分數的增大,脂溶性生物堿的提取率反而隨之下降。這可能是因為細胞在超聲破碎后,內容物釋放到溶液中,脂溶性生物堿與其他內容物之間在這個極性段共溶性較好。以體積分數75%和85%的乙醇作為提取溶劑,脂溶性生物堿的提取率都比55%的大,因此通過此單因素試驗,宜選擇體積分數65%、75%、85%的乙醇作為正交試驗中考察乙醇濃度影響的3個水平。
3.2.2超聲提取時間對脂溶性生物堿提取的影響 準確稱取5份臭常山粗粉各400 g,在料液比為1 g:10 mL、乙醇體積分數為65%的條件下,分別超聲提取40、50、60、80、100 min,按“2.5”節下的方法對各組脂溶性生物堿進行定性分析和定量測定,考察不同超聲提取時長對脂溶性生物堿提取率的影響,結果見圖7。
由圖7可知,臭常山脂溶性生物堿的提取率隨著超聲提取時間的延長而增大,但當時間長達80 min時,再延長時間脂溶性生物堿的提取率已變化不大,但超聲提取時長100 min,其提取率由80 min的0.288%增加到0.295%,提取率只提高了0.007百分點,分析其可能原因是在超聲進行到60 min時,細胞已經開始了大量破裂,細胞中所含生物堿大量釋放到提取液中。考慮到工藝要節能省時,故不考察超聲提取100 min后繼續延長超聲提取時長的影響,所以正交試驗在考察超聲提取時間對脂溶性生物堿提取率的影響時,只需考察60、80、100 min這3個水平。
3.2.3料液比對脂溶性生物堿提取的影響 準確稱取5份臭常山粗粉各400 g,在乙醇體積分數為65%的條件下,分別以料液比1:5、1:10、1:15、1:20、1:25(g:mL),超聲提取60 min,按“2.5”節下的方法對各組脂溶性生物堿進行定性分析和定量測定,考察不同料液比對脂溶性生物堿提取率的影響,結果見圖8。
由圖8可知,超聲提取過程中,脂溶性生物堿的提取率隨著提取過程中乙醇用量的增加而增大,當料液比為1 g:15 mL時,隨著乙醇用量的增加,脂溶性生物堿的提取率提高并不大,這是因為大部分細胞已經破碎,細胞中脂溶性生物堿都已釋放到溶液中,考慮到乙醇用量的增大會增加耗能和增長旋轉蒸發儀減壓濃縮浸膏的時間,耗能耗時,不符合工藝優化的目的。因此通過此單因素試驗,可以確定正交試驗考察料液比對脂溶性生物堿提取率的影響時,料液比的選擇范圍應在1 g:15 mL左右。
3.2.4超聲提取次數對脂溶性生物堿提取的影響 準確稱取臭常山粗粉400 g,在料液比為1 g:10 mL、乙醇體積分數65%的條件下,超聲提取60 min/次,分別提取1、2、3、4次,按“2.5”節下的方法對各組脂溶性生物堿進行定性分析和定量測定,分別考察超聲提取次數對脂溶性生物堿提取效果的影響,試驗結果見圖9。
由圖9可知,隨著超聲提取次數的增加,脂溶性生物堿的提取率亦在增大,當超聲提取進行1次時,脂溶性生物堿的提取率為0.28%,過濾后的藥渣進行與第1次相同條件的重復提取,與第2次提取率相比,第3次提高了0.04百分點;第4次比第3次只提高了0.01百分點,所以在優化臭常山脂溶性生物堿提取工藝過程中,在考察了乙醇濃度、超聲時間、料液比3個因素的正交試驗后,將得到優方案工藝進行檢驗,最后將優方案工藝進行3次超聲提取,即可達到試驗的工藝優化研究目的。
3.3脂溶性生物堿提取的正交試驗
在考察了上述單因素試驗的基礎上,臭常山脂溶性生物堿正交試驗設計要考察的各因素與水平詳見表2,正交試驗結果及分析見表3。
從表3可知,影響臭常山脂溶性生物堿提取率的各因素的主次順序依次為:乙醇體積分數>超聲提取時間>料液比,較優方案是用體積分數為65%的乙醇、在料液比1 g:20 mL的條件下超聲提取80 min。因為此方案在正交試驗中沒有,故需進行補充試驗進行驗證。按照優方案設計試驗,超聲提取3次,結果得到臭常山脂溶性生物堿的提取率為0.385%。
4討論與結論
與一般文獻報道的生物堿提取工藝不同,由于中藥材植物的理化特征容易受到生長地區、季節、年齡的影響,在進行有效成分分離提取工藝前,首先對試驗材料臭常山提取液進行HPLC、TLC指紋圖譜和水分、總灰分、醇浸出物含量等理化特征研究,其水分、總灰分、醇浸出物含量均值分別為8.21%、6.22%、6.20%,水分含量和醇浸出物含量測定的穩定性和重復性良好,可能是受取樣量和試驗精度要求的影響,醇浸出物含量測定的穩定性要稍差些。
與關于臭常山所含化學成分的分離鑒定文獻報道不同,本研究重點在臭常山的理化特征和脂溶性生物堿的提取工藝,影響其提取因素的順序依次為:乙醇體積分數>超聲提取時問>料液比;正交試驗確定的最佳工藝條件為:65%乙醇作為提取劑,料液比1 g:20 mL,超聲提取時間80 min,提取3次,脂溶性生物堿提取率為0.385%。富集到的脂溶性生物堿抗肺炎球菌、金黃色葡萄球菌及其他抑菌活性可進行深入研究。
