基于miRNA-靶位點配對的序列特征研究
2017-06-07 08:23:51滕少華夏飛迪劉冬寧鄒小勇
分析測試學報 2017年5期
滕少華,夏飛迪,張 巍,劉冬寧,王 洋,鄒小勇*
(1.廣東工業(yè)大學 計算機學院,廣東 廣州 510006;2.中山大學 化學學院,廣東 廣州 510275)
基于miRNA-靶位點配對的序列特征研究
滕少華1,夏飛迪1,張 巍1,劉冬寧1,王 洋2,鄒小勇2*
(1.廣東工業(yè)大學 計算機學院,廣東 廣州 510006;2.中山大學 化學學院,廣東 廣州 510275)
miRNA與其靶基因的作用機制十分復雜,因此,miRNA靶基因識別問題一直是miRNA研究領域的熱點難題。該文基于CLASH數(shù)據(jù)集,提出了miRNA-靶位點配對序列特征,并使用隨機森林建模。實驗結果表明,本模型的Acc,Sen,Spe,Pre以及Mcc分別達到90.05%,89.47%,90.56%,90.43%和0.799 8;ROC和PRC的AUC分別為0.954,0.958。相比已有方法,該方法表現(xiàn)出更加良好的性能,說明新引入的miRNA-靶位點配對序列特征對miRNA靶基因識別有很重大的影響。
miRNA;靶基因識別;CLASH;序列分析
MicroRNAs(miRNAs)是一類內源性、長約23個核苷酸(nt)的非編碼RNA,主要通過與mRNA 的3'UTR序列實現(xiàn)完全或不完全堿基互補配對,從而達到裂解mRNA和抑制mRNA翻譯成蛋白質的目的,在后轉錄時期和翻譯等級發(fā)揮重要的基因調節(jié)作用[1]。迄今已發(fā)現(xiàn)了2 000多個人類miRNA[2],這些miRNA可能調控著人體80%的基因,在各種生命活動和疾病調控中起著非常關鍵的作用[3]。由于miRNA靶基因識別的具體機制尚不明確,有效識別miRNA靶基因成為miRNA研究領域的一大難題。單純用蛋白免疫印跡法[4]以及微陣列法等[5]生物實驗方法來鑒別miRNA靶基因,費時而且耗費大。因此借助化學生物信息方法,挖掘miRNA潛在的靶基因,能進一步探討miRNA作用機制和miRNA基因調控網絡,具有重要理論意義和實用價值。
近十年來,研究者提出了多種生物計算方法識別miRNA靶基因。Enright 等[6]通過給miRNA與其靶基因的配對情況進行打分,計算miRNA與靶基因形成雙鏈后的最小自由能,同時引入靶位點保守性作為最后一個條件,經過層層篩選,得到潛在的miRNA靶基因。Lewis等[7]提出了“種子”區(qū)(miRNA 5'端開始第 2到第 8位核苷酸的區(qū)間)的概念,發(fā)現(xiàn)種子區(qū)域的匹配情況對miRNA靶基因的識別有重大影響。Kertesz等[8]考慮了靶基因的二級結構,提出靶位點可接性概念,認為miRNA與靶基因的結合能力會受到不同二級結構影響。作為第一代生物計算方法,盡管研究人員發(fā)現(xiàn)了較多有用的特征,但研究表明[9-10],這些特征并不完全適用于miRNA與靶基因結合的情況。將這些特征作為篩選條件,會大大提高假陰性的預測率,于是基于機器學習的第二代生物計算的方法應運而生。
用機器學習的方法預測miRNA靶基因,其基本原理是采用可靠的數(shù)據(jù)集,根據(jù)所提出的特征,將miRNA和靶基因的結合序列特征數(shù)字化,然后將這些特征加以融合對所構建的模型進行訓練,并對靶基因進行預測。Huang等[11]從表達圖譜數(shù)據(jù)中提取樣本用于訓練模型,文獻[12-14]使用交聯(lián)和免疫沉淀技術(CLIP)數(shù)據(jù)用于模型訓練。最近,Helwak等[15]提出的CLASH(crosslinking ligation and sequencing of hybrids)直接提供了miRNA和其對應的靶位點序列數(shù)據(jù),為進一步研究miRNA與其靶基因位點序列作用提供了良好的平臺。近年來,許多報道采用miRNA與靶位點形成雙鏈的最小自由能,miRNA種子區(qū)域的配對數(shù)目,靶位點保守性,以及靶位點的可接入性等常用的特征[16-20]進行研究,但這些方法均有特異性太低的缺點。因此構建miRNA與靶基因結合特征,對miRNA靶基因識別具有重要意義。
本文基于CLASH實驗數(shù)據(jù)集,提出了基于miRNA-靶位點配對的特征,結合一系列傳統(tǒng)特征,運用隨機森林算法建立模型,進行了miRNA靶基因識別,并與文獻報道的其它兩個采用同樣數(shù)據(jù)集建立的模型進行了比較。
1 實驗部分
1.1 數(shù)據(jù)提取與處理
在CLASH實驗中提取了18 514條數(shù)據(jù),其中包含399個miRNAs和18 514個靶位點數(shù)據(jù),并將這些樣本數(shù)據(jù)全部作為正樣本,每一條樣本中均包括miRNA和靶位點信息。由于負樣本匱乏,將CLASH數(shù)據(jù)集中所有的miRNAs和靶位點進行一一配對,共有3 693 543個數(shù)據(jù)組合。然后去除其中的正樣本,按正負樣本1∶1比例,從3 675 029條數(shù)據(jù)中隨機選擇18 514條數(shù)據(jù)作為本研究所需的負樣本。為了研究隨機樣本對所構建模型的影響,進行了多次重復實驗來檢測模型的魯棒性。
1.2 特征集合
共選用26種特征(84個特征值),具體的特征集合如表1所示。其中前21種特征共57個特征值,文獻[16-20]已有報道;本文所構建的后5種特征(ID 22~26),含有27個特征值,這些特征值充分考慮了miRNA與其靶基因的作用情況。
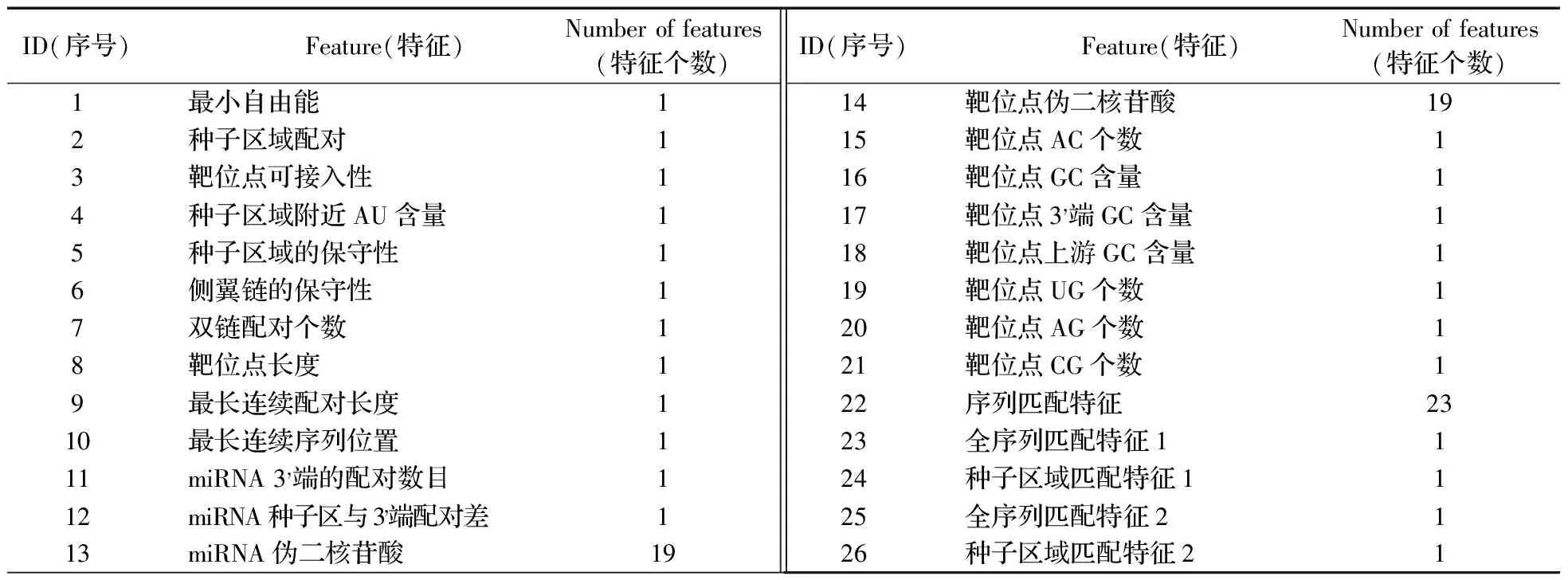
表1 miRNA與靶位點結合特征集合Table 1 Feature sets of the interaction of miRNA and target sites
1.3 特征選擇
特征選擇針對高維度數(shù)據(jù)計算問題而提出,通過剔除冗余特征和無關特征,提高機器學習算法的泛化性能和運行效率。本文使用最小冗余最大相關算法(Minimal redundancy maximal relevance criterion,mRMR)[21]對84個特征排序,并選擇了最優(yōu)特征子集構建模型。
1.4 隨機森林
隨機森林[22]是一種組合方法,由許多的決策樹組成,因這些決策樹的形成采用了隨機的方法,因此也稱作隨機決策樹。隨機森林中的樹之間沒有關聯(lián),當測試數(shù)據(jù)進入隨機森林時,讓每一棵決策樹進行分類,最后取所有決策樹中分類結果最多的那類為最終結果。本文使用隨機森林機器學習方法作為訓練模型,算法來源于Scikit-learn(http://scikit-learn.org/stable/)工具包[23],整個程序使用python開發(fā),并優(yōu)化了森林中樹的數(shù)目和每棵樹的特征數(shù)兩個參數(shù)。
1.5 性能指標
分類器的性能可通過一些獨立的指標進行評估。為評估模型的性能,以準確率(Acc)、敏感度(Sen)、特異性(Spe)、精確度(Pre)和馬氏相關系數(shù)(Mcc) 5種指標來評估模型的性能。這些指標的計算方法如下:
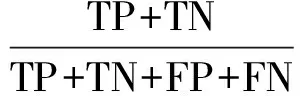
其中,TP指被判定為正樣本,也是正樣本的數(shù)目;TN指被判定為負樣本,也是負樣本的數(shù)目;FN指被判定為負樣本,但事實上是正樣本的數(shù)目;FP指被判定為正樣本,但事實上是負樣本的數(shù)目。此外,受試者工作特征曲線(Receiver operating characteristic curve,ROC曲線)和 準確率-召回率曲線(Precision-recall curve,PRC曲線)也用于評估模型的性能。ROC曲線是反映敏感性和特異性連續(xù)變量的綜合指標,采用構圖法揭示敏感性和特異性的相互關系,將連續(xù)變量設定出多個不同的臨界值,從而計算出一系列敏感性和特異性。再以敏感性為縱坐標、(1-特異性)為橫坐標繪制成曲線,曲線下面積越接近1,表明模型性能越好。PRC曲線是反映準確率和召回率(敏感性)連續(xù)變量的綜合指標,采用構圖法揭示準確率和敏感性的相互關系,將連續(xù)變量設定出多個不同的臨界值,從而計算出一系列準確率和敏感性。再以準確率為縱坐標、敏感性為橫坐標繪制成曲線,曲線下面積越接近1,表明模型性能越好。
1.6 實驗流程
miRNA的靶基因預測屬于機器學習問題,整個實驗的流程如圖1所示。
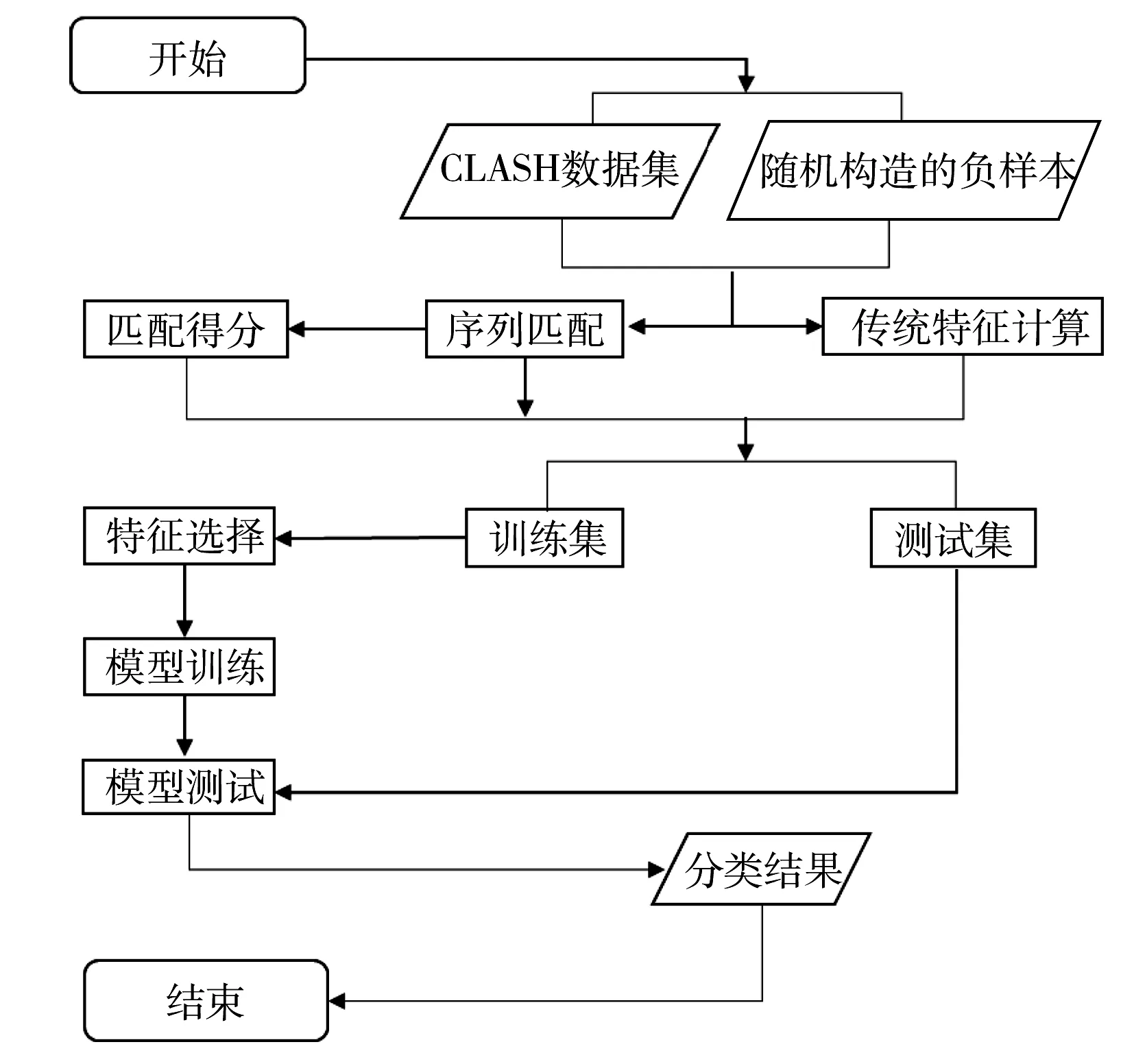
圖1 實驗流程圖Fig.1 The flow chart of the experiment
步驟1:選擇CLASH數(shù)據(jù)集作為正樣本,并根據(jù)該數(shù)據(jù)集構造負樣本,將CLASH數(shù)據(jù)集中的miRNA與靶位點序列隨機配對,刪除其中的正樣本,再從剩余的數(shù)據(jù)集中隨機選擇18 514條作為負樣本,正負樣本比例為1∶1;
步驟2:根據(jù)傳統(tǒng)特征的計算方法,計算樣本傳統(tǒng)特征的特征值;
步驟3:采用改進的Smith-Waterman方法將正負樣本進行序列匹配,并轉換為二進制序列。再根據(jù)正樣本序列匹配的情況構造權重向量W,并以此向量計算正負樣本的序列匹配得分特征;
步驟4:采用隨機森林的方法構建模型,并訓練模型的參數(shù);
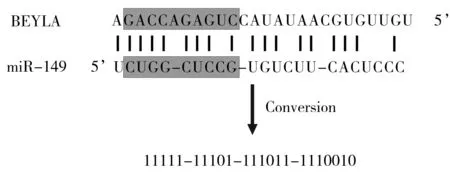
圖2 序列匹配二進制化表示Fig.2 A binary representation of sequence match
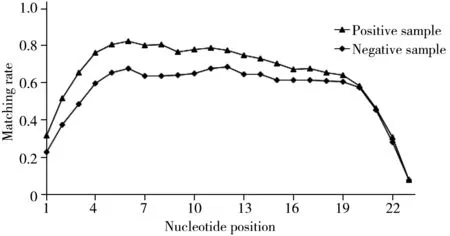
圖3 正負樣本匹配對比Fig.3 Comparison of positive and negative samples matching
步驟5:模型測試;
步驟6:與其他模型比較并分析。
2 結果與討論
2.1 基于miRNA-靶位點的配對miRNA與其靶位點并不完全匹配,且匹配情況差異很大。本文根據(jù)樣本集中miRNA與其靶位點的配對情況,將每一條miRNA與靶位點結合后的雙鏈表示為由“0”和“1”組成的二進制序列,并對構成的二進制序列進行分析,具體過程如圖2所示,其中陰影部分為“種子區(qū)域”。
在圖2中,BEYLA序列為miR-149對應的靶位點序列。首先采用改進的Smith-Waterman方法[24],按照堿基A∶U和G∶C互補配對原則進行序列匹配,允許G∶U錯配。從miR-149序列5'端的第1個核苷酸開始,與BEYLA序列的每1個核苷酸進行比對,如果匹配,則用“1”表示,對應的核苷酸在其相應的位置用豎線“|”連接;如果不匹配,則用“0”表示。每1條序列中均可能有一些短橫線“-”,表示該位置不含任何核苷酸。因此,miR-149序列和BEYLA靶位點序列的匹配可轉化為二進制序列“11111111011110111110010”,共含有23個“0”和“1”特征值。因為CLASH數(shù)據(jù)集中大部分miRNA的長度為23,因此本文將每一條miRNA與靶位點結合后的雙鏈轉換為23個“0”或“1”組成的特征值序列,如果miRNA的長度小于23,則該特征值用0補充,如果miRNA長度大于23,多出來的特征值不予考慮。最后,本文將這23個特征值加入特征集。
采用以上數(shù)字編碼的方法,對CLASH數(shù)據(jù)集和隨機構造的負樣本進行了比對分析。首先,將每一個樣本進行了序列匹配,然后轉換為二進制“0”和“1”序列,并統(tǒng)計每個位置配對成功的概率,結果如圖3所示。
在圖3中,橫軸表示miRNA每個核苷酸的位置,縱軸表示miRNA上每個位置配對成功的概率。圖中上面的曲線表示正樣本中miRNA上每個位置配對成功的概率,下面的曲線表示負樣本中miRNA上每個位置配對成功的概率。從圖3可以發(fā)現(xiàn),正樣本整體的匹配情況比負樣本好,特別是第20位之前的匹配情況,正樣本明顯優(yōu)于負樣本。同時實驗還發(fā)現(xiàn),正負樣本序列兩端配對成功的概率遠遠低于中間核苷酸位置配對成功的概率。本文對每一個位置的差異值進行計算,結果發(fā)現(xiàn),正負樣本在第2到第8位的配對情況的差異相對于其他位置大很多,這與之前的研究觀點一致[25],即miRNA種子區(qū)域的配對情況對miRNA的靶基因識別具有很重要的作用。
基于上述發(fā)現(xiàn),本文根據(jù)正樣本中每個位置的匹配成功率構造了一個權重向量w,并以此向量為基礎,提出了幾種方法對miRNA的匹配序列進行打分,得到了4個關鍵特征。
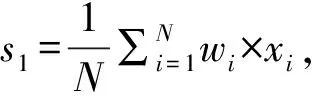
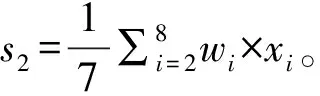
“全序列匹配特征1” 和“種子區(qū)域匹配特征1”考慮了配對成功對miRNA靶基因識別的影響。
特征3:對于miRNA上第i位的匹配情況xi=1,如果xi=1,其對應的權值為wi;如果x1=0,其對應的權值則為qi=1-wi,構建了“全序列匹配特征2”,可以通過計算整段序列匹配得分的平均值s3,其計算公式如下,其中N(N=23)為序列長度。
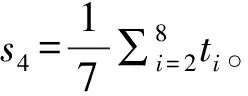
其中,特征3和特征4,既考慮了匹配成功情況,也考慮了匹配不成功的情況。因此,根據(jù)miRNA-靶位點配對情況,構建了23個序列特征和“全序列匹配特征1”、“種子區(qū)域匹配特征1”、“全序列匹配特征2” 和“種子區(qū)域匹配特征2”的4個序列得分特征,共27個特征值。
2.2 特征選擇
根據(jù)表1構建了包含84個特征的特征集,為了研究各特征的貢獻,采用mRMR方法對各特征進行排序,前29個特征排名如表2所示。由表2可見,所構建的“種子區(qū)域匹配特征1”排名第4,“全序列匹配特征1”排名第5,“種子區(qū)序列匹配特征2”排名第8,“全局序列匹配特征2”排名第9。說明新構建的特征對miRNA靶基因識別具有相當重要的作用。同時還可以看到,傳統(tǒng)特征如最小自由能、保守性和種子區(qū)域配對均對miRNA靶基因識別起著重要作用。
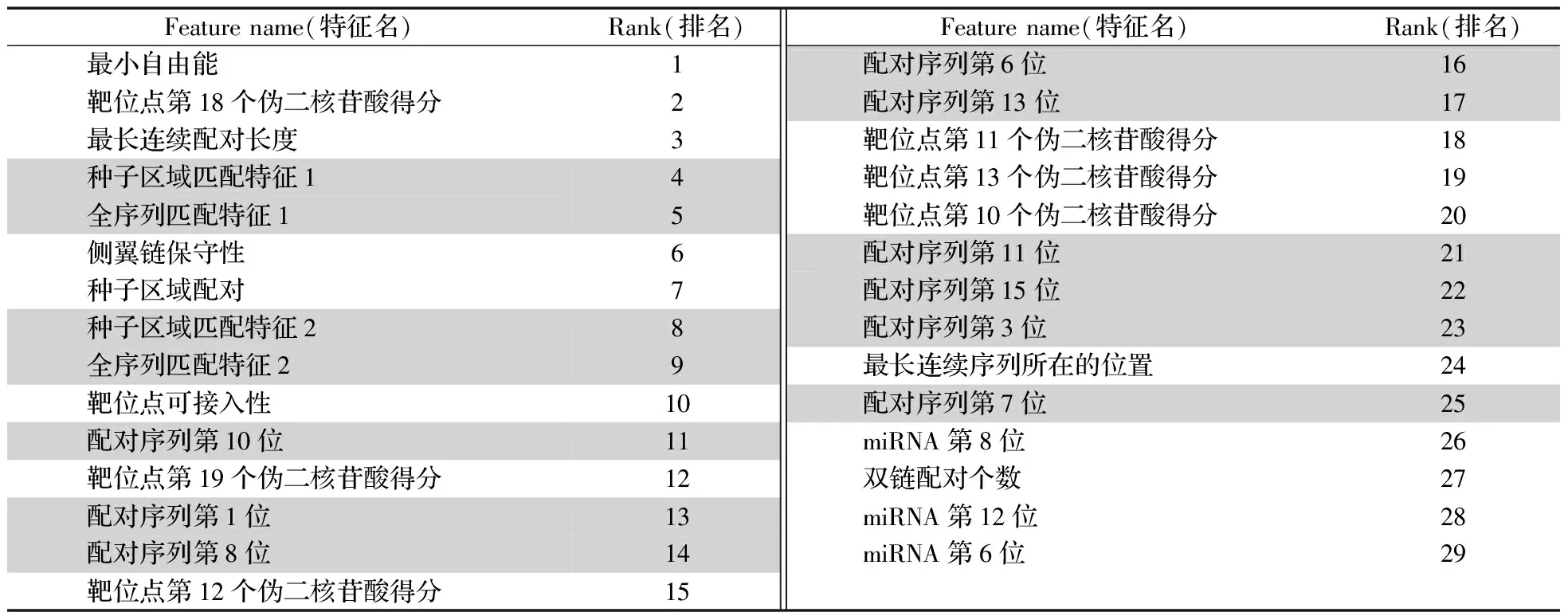
表2 前29個特征排名Table 2 The top 29 features sorted by mRMR
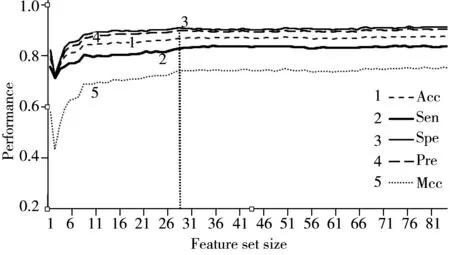
圖4 基于不同特征子集的預測結果Fig.4 The prediction results based on the various feature subset
根據(jù)各特征的排名,以1為梯度,分別使用排名前84,83,…,3,2,1個特征組成的特征子集,然后基于每一個特征子集構建對應的模型,計算了Acc,Sen,Spe,Pre以及Mcc,以考察所構建模型的性能,具體結果如圖4所示。從圖4可以看出,當特征子集中的特征數(shù)大于29時,模型性能基本無變化,因此本文最終選擇前29個特征作為特征子集。在排名前29個特征中,本文提出的特征共有13個(如表2的陰影所示),表明本文提出的特征是可行的。
2.3 參數(shù)訓練
隨機森林有兩個重要的參數(shù),n_estimators表示森林中樹的棵數(shù),max_feature表示每次生成決策樹時選擇的特征個數(shù)。對于n_estimators,以100梯度,提取100~1 000的所有取值(100,200,…,1 000)。對于max_feature,研究了scikitlearn軟件包中所有取值。結果表明,當n_estimators=400和max_feature=4時,模型的性能達到最佳。
2.4 魯棒性評估
依據(jù)上述步驟,建立了基于隨機森林算法的模型,對miRNA靶基因進行預測。為研究模型的魯棒性,將負樣本進行了10次隨機采樣,根據(jù)所建立的數(shù)據(jù)集,構建模型和計算各性能指標,具體結果如表3所示。
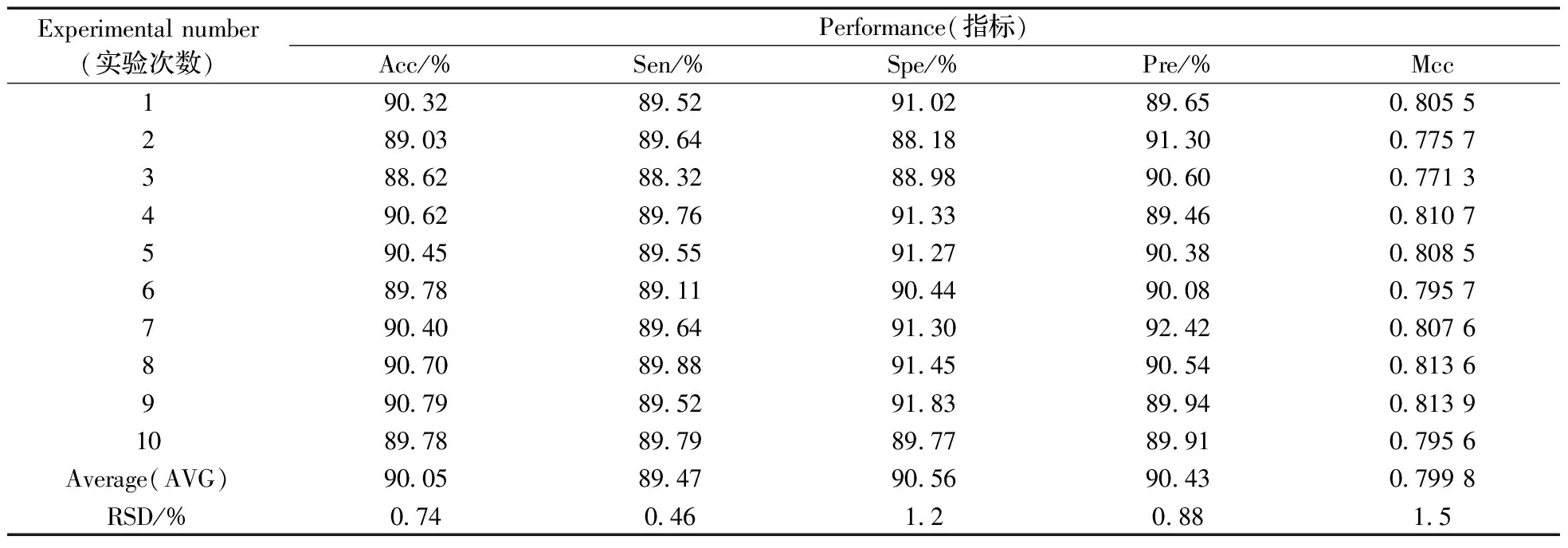
表3 模型魯棒性評估的結果Table 3 The results of the robustness of the model
從表3可以看出,準確率(Acc)、敏感度(Sen)、特異性(Spe)、精確度(Pre)、馬氏相關系數(shù)(Mcc)的平均值分別為90.05%,89.47%,90.56%,90.43%,0.799 8,而且RSD均小于1.6%。結果表明,本文所建立的模型具有很強的魯棒性。同時,基于最高的Acc值,本文還計算了模型的ROC和PRC值。結果顯示,該模型的ROC和PRC曲線下面積分別可達到0.953 7,0.958 4,說明模型對于靶基因預測表現(xiàn)出良好的性能。
2.5 與其他方法比較
為了驗證新構建特征的有效性,基于傳統(tǒng)特征集構建了miRNA靶基因預測模型,并與本文所使用的模型進行比較。結果表明,當加入新構建的特征后,模型的性能得到了很大的提高,Acc提高了6%,Spe和Pre均提高了近5%,Sen的改善明顯(提高了近9%),ROC和PRC的AUC提高了10%左右,進一步驗證了新構建特征的有效性。同時,將本方法與已有的MirTarget[26]和TarPmiR[27]方法進行比較(表4),可以看到,本文所采用的模型整體性能更好。其中本方法的Acc相比TarPmiR和MirTarget分別增加了8%和5%,實驗結果明顯改善。同時本模型的ROC和PRC的AUC高達0.95以上,也驗證了本模型性能的穩(wěn)定性。
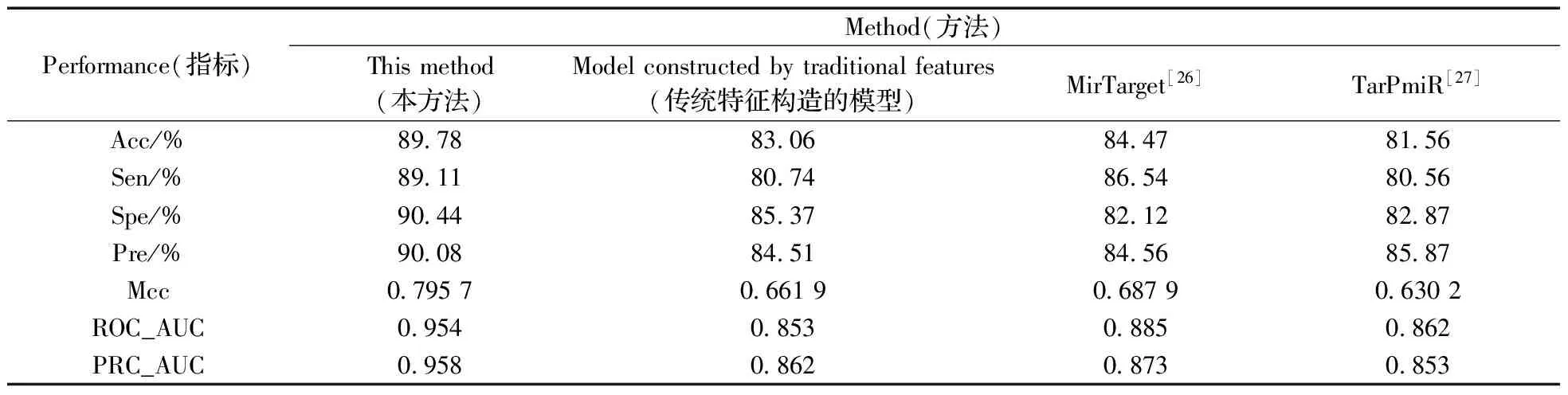
表4 不同方法的比較Table 4 The comparision of different method
3 結 論
本文基于CLASH數(shù)據(jù)集,構造了27個miRNA-靶位點配對序列相關特征,結合傳統(tǒng)特征,組成了1個包含84個特征值的特征集合,并使用隨機森林模型進行機器學習,構造miRNA靶基因預測模型。本模型的平均Acc,Sen,Spe,Pre以及Mcc分別達到90.05%,89.47%,90.56%,90.43%和0.799 8,ROC和PRC的AUC分別為0.954和0.958,表現(xiàn)出良好的預測性能。根據(jù)實驗結果可以預見,所建立的方法可為miRNA靶基因預測提供一定的技術支撐。
[1] Bartel D P.Cell,2004,116(2):281-297.
[2] Kozomara A,Griffithsjones S.NucleicAcidsRes.,2014,42(D1):68-73.
[3] Ambros V.Nature,2004,431(7006):350-355.
[4] Lagos Q M,Rauhut R,Lendeckel W,Tuschl T.Science,2001,294(5543):853-858.
[5] Lin L P,Lau N C,Garrett-Engele P,Grimson A,Schelter J M,Castle J,Bartec D P,Linsley P S,Johnson J M.Nature,2005,433(7027):769-773.
[6] Enright A J,John B,Gaul U,Tuschl T,Sander C.GenomeBiol.,2003,5(1):R1.
[7] Lewis B P,Shih I H,Jones-Rhoades M W,Bartel D P,Burge C B.Cell,2004,115(7):787-798.
[8] Kertesz M,Iovino N,Unnerstall U,Gaul U,Segal E.NatureGenetics,2007,39(10):1278-1284.
[9] Didiano D,Hobert O.Nat.Struct.Mol.Biol.,2006,13(9):849-851.
[10] Sethupathy P,Megraw M,Hatzigeorgiou A G.Nat.Methods,2006,3(11):881-886.
[11] Huang J C,Babak T,Corson T W,Chua G,Khan S,Gallie B L,Hughes T R,Blencowe B J,Frey B J,Morris Q D.Nat.Methods,2007,4(12):1045-1049.
[12] Liu C,Mallick B,Long D,Rennie W A,Wolenc A,Carmack C S,Ding Y.NucleicAcidsRes.,2013,41(14):338-345.
[13] Reczko M,Maragkakis M,Alexiou P,Grosse I,Hatzigeorgiou A G.Bioinformatics,2012,28(6):771-776.
[14] Gumienny R,Zavolan M.NucleicAcidsRes.,2015,43(3):1380-1391.
[15] Helwak A,Kudla G,Dudnakova T,Tollervey D.Cell,2013,153(3):654-665.
[16] Yousef M,Jung S,Kossenkov A V,Showe L C,Showe M K.Bioinformatics,2007,23(22):2987-2992.
[17] Wang X.Bioinformatics,2014,30(10):1377-1383.
[18] Shuang C,Guo M,Wang C,Wang C,Liu C Y,Liu Y.IEEE/ACMTrans.Comput.Biol.Bioinf.,2015,13(6):1145-1169.
[19] Yu S,Kim J,Min H,Yoon S.Methods,2014,69(3):220-229.
[20] Bandyopadhyay S,Mitra R.Bioinformatics,2009,25(20):2625-2631.
[21] Ponsa D,López A.IberianConferenceon-PatternRecognitionandImageAnalysis,2007,4477:47-54.
[22] Liaw A,Wiener M.RNews,2001,2(3):18-22.
[23] Pedregosa F,Varoquaux G,Gramfort A,Michel V,Thirion B,Grisel O,Blondel M,Prettenhofer P,Weiss R,Dubourg V,Vanderplas J,Passos A,Cournapeau D,Brucher M,Perrot M,Duchesnay E.J.Mach.Learn.Res.,2013,12(10):2825-2830.
[24] Smith T F,Waterman M S,Fitch W M.J.Mol.Evol.,1981,18(1):38-46.
[25] Agarwal V,Bell G W,Nam J W,Bartel D P.Elife,2015,4:e05005.
[26] Wang X.Bioinformatics,2016,32(9):1316-1322.
[27] Ding J,Li X M,Hu H Y.Bioinformatics,2016,32(18):2768-2775.
Study of Sequence Features Based on MicroRNA-Target Sites Pairing
TENG Shao-hua1,XIA Fei-di1,ZHANG Wei1,LIU Dong-ning1,WANG Yang2,ZOU Xiao-yong2*
(1.Faculty of Computer,Guangdong University of Technology,Guangzhou 510006,China;2.School of Chemistry, Sun Yat-sen University,Guangzhou 510275,China)
The mechanisms underlying the interaction of miRNAs with their mRNA targets are quite complex,which makes miRNA target prediction be a hot issue in the field of miRNA research.The features of miRNA-target sites pairing sequences were proposed based on the CLASH dataset,and the random forest models were applied for modeling.The average values of Acc,Sen,Spe,Pre and Mcc are 90.05%,89.47%,90.56%,90.43% and 0.799 8,respectively,and the AUC of ROC and PRC are 0.954 and 0.958,respectively.The results indicated that the current method shows a better performance compared with the existed methods, and the features newly constructed have a very significant impact on the identification of miRNA target genes.
miRNA;target prediction;CLASH;sequence analysis
2016-12-12;
2017-01-20
國家自然科學基金項目(21675180);廣東省科技計劃項目(2014A040401022,2015A030401033,2016B010108007);廣州市科技計劃項目(201604020145)
10.3969/j.issn.1004-4957.2017.05.006
O629.74;TB9
A
1004-4957(2017)05-0614-07
*通訊作者:鄒小勇,博士,教授,研究方向:化學計量學、電分析化學,Tel:020-84114919,E-mail:ceszxy@mail.sysu.edu.cn
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
中學生數(shù)理化·七年級數(shù)學人教版(2020年10期)2020-11-26 08:24:50
數(shù)學物理學報(2020年2期)2020-06-02 11:29:24
瘋狂英語·新策略(2019年10期)2019-12-13 08:43:28
當代陜西(2019年10期)2019-06-03 10:12:04
數(shù)學小靈通·3-4年級(2017年9期)2017-10-13 08:10:54
光學精密工程(2016年6期)2016-11-07 09:07:19
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
