三唑噻吩并嘧啶衍生物殺菌活性的QSAR研究
2017-06-10 18:16:53岳瑋馮惠馮長君
湖北農業科學 2017年9期
岳瑋++馮惠++馮長君
摘要:為了研究三唑噻吩并嘧啶衍生物對小麥赤霉菌抑菌活性(G/%)的定量構效關系(QSAR),按照分子的拓撲環境編程計算了14種上述化合物的電性距離矢量(MD)。通過最佳變量子集回歸,建立了它們的二參數(M26、M32)QSAR模型,非交叉驗證相關系數(R2)與逐一剔除法交叉驗證相關系數(Rcv2)分別為0.857、0.648,顯示良好的穩健性和預測能力。根據進入模型可知,影響三唑噻吩并嘧啶衍生物對小麥赤霉菌抑菌活性的主要因素是>C-、-O等結構碎片。
關鍵詞:三唑噻吩并嘧啶衍生物;小麥赤霉菌;抑菌活性;電性距離矢量;定量構效關系
中圖分類號:S482.2;O6-051 文獻標識碼:A 文章編號:0439-8114(2017)09-1665-03
DOI:10.14088/j.cnki.issn0439-8114.2017.09.016
Study on QSAR of the Bactericidal Activity for 1,2,4-Triazole-Thieno[2,3-d] Pyrimidin Derivatives to Gibberella
YUE Wei, FENG Hui, FENG Chang-jun
(School of Chemistry & Chemical Engineering, Xuzhou Institute of Technology, Xuzhou 221111, Jiangsu, China)
Abstract: To study the quantitative structure-activity relationship(QSAR) of the bactericidal activity(G/%) for 1,2,4-triazole-thieno[2,3-d] pyrimidin derivatives to wheat gibberella, the molecular electronegativity distance vector(MD) of 14 above compounds were calculated by program according to molecular topological environment. The two-parameter(M26, M32) QSAR model for the compounds was constructed by leaps-and-bounds regression(LBR). The traditional correlation coefficient (R2) and cross-validated coefficient of multiple determination(Rcv2) of leave-one-out(LOO) were 0.857 and 0.648 respectively, which demonstrated good robustness and predictive ability of the model. From the two parameters of the model, it was known that the dominant influence factors of the bactericidal activity were the molecular structure fragments:>C-,O- in the derivatives.
Key words: 1,2,4-triazole-thieno[2,3-d] pyrimidin derivative; wheat gibberella; bactericidal activity; electronegativity distance vector; quantitative structure-activity relationship
噻吩并嘧啶衍生物具有多變的化學結構, 表現出良好的抗菌、抗病毒、抗腫瘤、消炎、抗過敏、抗瘧疾等作用[1-3]。周新等[4]、唐傳球等[5]為了篩選出更好的1,2,4-三唑并[1,5-a]嘧啶類殺菌劑,采用活性單元拼接法在1,2,4-三唑并[1,5-a]嘧啶環上分別引入芐基硫醚和不同烷氨基等,設計并合成了14個新型的5-甲基-1,2,4-三唑噻吩并[1,5-a]嘧啶類衍生物,簡稱“三唑噻吩并嘧啶衍生物”,并測試了它們對小麥赤霉菌(wheat gibberella)的抑菌活性(G/%)。為了探討影響這些化合物抑菌活性的分子結構基團,本研究基于Liu等[6,7]、Zhang等[8]的電性距離矢量(Molecular electronegativity distance vector,簡稱Md)與它們殺菌活性進行構效關系(Quantitative structure-activity relationship,QSAR)[9-12]研究,以便獲得此類結構新穎、高效低毒農藥提供理論指導。
1 材料與方法
1.1 三唑噻吩并嘧啶衍生物的分子結構與抑菌活性
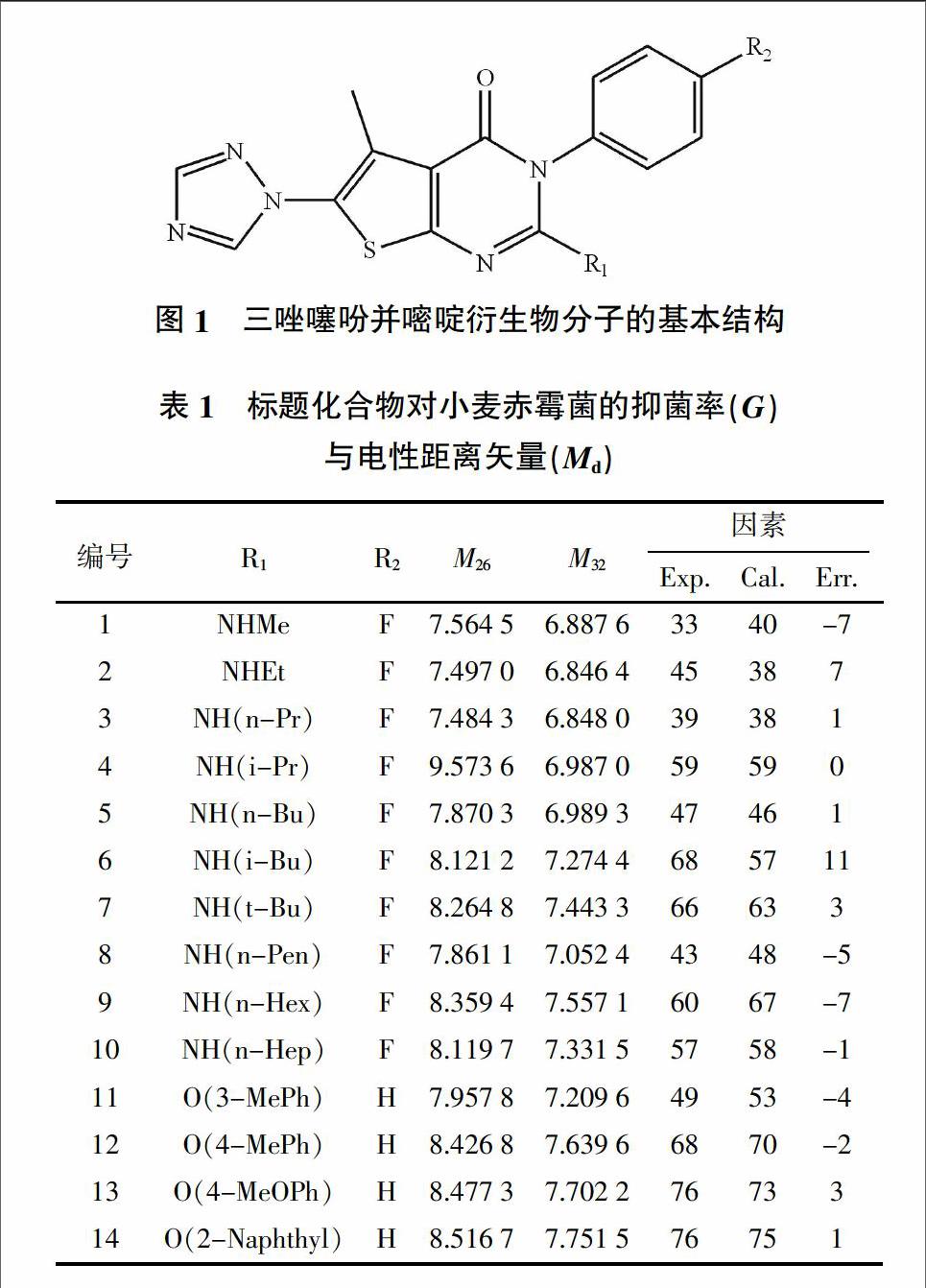
周新等[4]、唐傳球等[5]合成的14個三唑噻吩并嘧啶衍生物分子的基本結構見圖1,其中的取代基(R1,R2)見表1。將這些化合物配制成濃度為5×10-5 g/L的試樣,采用含毒介質法對小麥赤霉菌進行抑菌活性測試。抑菌活性以抑菌率(G/%)表示,G值越大,相應化合物殺菌能力越強。
1.2 構建電性距離矢量
拓撲指數衍生于分子隱氫圖的各種數學矩陣(如距離矩陣、鄰接矩陣等),用于揭示分子結構某種特征的描述符,無須試驗測定,被廣泛應用于物質構效關系(QSAR)研究中。Liu等[6,7]提出的電性距離矢量(Md)是根據每一個非氫原子在分子中的拓撲環境以及成鍵的電子信息進行矩陣運算得到的多組數值,以表達分子中的所有非氫原子的電性特征和拓撲特征的相互作用。其拓撲性是基于原子之間的圖論距離或拓撲距離,電子性質則基于該非氫原子固有狀態以及分子中其他原子對此固有狀態的影響。分子電性距離矢量的構建可分為以下三步:
首先是給非氫原子i分類,相應的固有狀態值(Ii)為:
Ii=(mi/4)0.5[(2/Ni)2δiv+1]/δi (1)
式中,mi為該非氫原子i價電子數,Ni為該非氫原子i價電子的主量子數,δi與δiv為其點價及支化度。由于分子中,每一個原子都會受到其他原子的干擾。因此,每種非氫原子的相對電性(qi)定義為:
qi=Ii+ΔIi=Ii+Σ(Ii-Ij)/fij2 (2)
式中,ΔIi是分子中其他所有非氫原子對原子i的擾動之和,fij為分子圖中原子i和j間邊的數值。最后將所有非氫原子的相對電性之間的相互作用之和定義為電性距離矢量(Mk,l=Md):
Mk,l=Md=Σ(qi×qj)/fij2 (3)
式中,k,l=1-13;w=1-91。由此可見,Md修正了多種著名拓撲指數的局限,能夠較為全面反映分子的拓撲、幾何及電性特征的分子電性距離矢量。本研究中三唑噻吩并嘧啶衍生物分子中涉及9種原子類型(-CH3、-C-、-C<、>C<、-N-、>N-、-O、-O-、-S-、X-),它們兩兩組合,理論上可以形成45種電性距離矢量。實際只有39種,原因是它們在每個分子中并不是都存在。本研究中三唑噻吩并嘧啶衍生物的部分電性距離矢量見表1。
1.3 統計分析方法
將每種三唑噻吩并嘧啶衍生物的39種電性距離矢量與相應的抑菌活性(G)構建數據集,應用最佳子集回歸選擇最佳變量組合,建立G的定量結構-生物活性相關性(QSAR)模型,采用逐一剔除法(LOO)對模型的預測能力及穩健度進行檢驗,以交叉驗證相關系數(Rcv2)予以評價。LOO法的基本思想是從研究總體中每次只剔除一個化合物,用余下化合物的數據建立模型,以此模型對被剔除化合物予以預測;并以預測值關聯試驗值,得交叉驗證系數(Rcv2)。一般公認Rcv2≥0.5,所建模型具有良好的魯棒性與預測能力[13]。在多元回歸分析中,為使所得預測模型具有較高的可信度,一般遵循如下經驗規則:f/b≥5[14];式中,b是引進回歸模型中自變量的個數。
2 結果與討論
2.1 三唑噻吩并嘧啶衍生物抑菌活性與Md的QSAR模型
經最佳子集變量回歸選擇標題化合物分子的39個電性距離矢量與其G的合適變量組合,所得二元最佳QSAR模型:
G=-235.299+8.008M26+31.191M32 (4)
f=14,R2=0.857,Radj2=0.831,F=33.052,S=5.639,P=0.000
式中,f、R2、Radj2、Rcv2、S、F分別為樣本容量、判定系數(亦稱“削減誤差比例”)、校正判定系數(以消除自變量個數及樣本容量對判定系數的影響)、估計標準誤差、Fischer檢驗值。將數據M26、M32帶入模型(4)中,其計算值(見表1中“Cal.”)與相應試驗值基本吻合。
2.2 模型(4)的質量檢驗
模型(4)的f/b=7>5,說明該模型具有統計學意義。通常用方差膨脹因子(Variance inflation factors,VIF)[15]評價模型中各自變量的多重相關性,其定義式為:VIF=1/(1-β2) (5)
式中,β2為進入模型的所有自變量中某一個自變量與余下自變量的判定系數。如VIF=1,表明各自變量間完全不相關;當VIF<5時,說明變量間沒有明顯的自相關性;當VIF>5時,說明變量間存在明顯的共線性,所建模型不能用于估算與預測。式(4)中,M26、M32的VIF依次為1.252、1.252,非常接近于1,表明兩者之間幾乎不相關,證明該模型中不存在共線性,是高度穩定的。模型(6)的Rcv2=0.648≥0.5,所建模型具有良好的魯棒性與預測能力。
2.3 三唑噻吩并嘧啶衍生物的QSAR模型分析
進入模型(4)中的電性距離矢量是:M26反映第三類碳原子(-C<,包含共軛體系,下同)與第三類碳原子(-C<)相互作用,M32反映第三類碳原子(-C<)與第九類氧原子(-O,實指酮基氧)相互作用。模型(4)中M26和M32前的系數均大于零,表明在母體結構(圖1)相同下,分子中包含第三類碳原子與第九類氧原子越多,其M26和M32值越大,相應化合物的抑菌活性越強。模型(4)的削減誤差比例R2為0.857,只有近14.3%的未知因素未被揭示,這也表明M26、M32與常數項共同揭示了影響三唑噻吩并嘧啶衍生物對小麥赤霉病菌殺菌活性的本質因素。
3 結論
本文基于電性距離矢量,通過多元統計分析研究14種三唑噻吩并嘧啶衍生物對小麥赤霉病菌殺菌活性。
1)所建模型(4)具有統計學意義,經Rcv2、VIF等檢驗,表明該模型具有良好的穩健性及預測能力。
2)模型(4)的削減誤差比例R2為0.857,結果表明:①M26、M32與常數項共同揭示了影響三唑噻吩并嘧啶衍生物對小麥赤霉病菌殺菌活性的本質因素;②在母體結構相同下,影響三唑噻吩并嘧啶衍生物對小麥赤霉病菌殺菌活性的主要因素是其分子的二維結構:-C<、-O等結構碎片。
參考文獻:
[1] 胡海軍,嚴 凱,陳 鴻,等.噻吩并嘧啶酮衍生物的合成及其抑菌活性[J].化學研究與應用,2012,24(1):31-36.
[2] 陳 玉,柏 舜,賀紅武,等.新型四氫苯并[4,5]噻吩并[2,3-d]嘧啶酮衍生物的合成及其抗腫瘤活性[J].有機化學,2014,34(11):2362-2369.
[3] 曾國平,鄭 平.哌啶并噻吩并嘧啶酮衍生物的合成、表征及生物活性[J].化學學報,2012,70(6):759-764.
[4] 周 新,孫 勇.2-烷氨基-3-對氟苯基-5-甲基-6-(1H-1,2,4-三唑-1-基)-噻吩并[2,3-d]嘧啶-4(3H)-酮衍生物的合成及其殺菌活性[J].化學試劑,2015,37(4):361-364.
[5] 唐傳球,孫 勇,章平平.2-芳氧基-3-苯基-5-甲基-6-(1H-1,2,4-三唑-1-基)噻吩并[2,3-d]嘧啶-4(3H)-酮衍生物的合成及其抑菌活性研究[J].長江大學學報(自科版),2015,12(28):47-50.
[6] LIU S S,YIN C S,LI Z L,et al. QSAR study of steroid benchmark and dipeptides based on MEDV-13[J].J Chem Inf Comput Sci,2001,41(2):321-329.
[7] LIU S S,YIN C S,WANG L S. Combined MEDV-GA-MLR method for QSAR of three panels of steroids,dipeptides,and COX-2 inhibitors[J].J Chem Inf Comput Sci,2002,42(3):749-756.
[8] ZHANG Y H,XIA Z N,QIN L T,et al. Prediction of blood-brain partitioning:A model based on molecular electronegativity distance vector descriptors[J].Journal of Molecular Graphics and Modelling,2010,29(2):214-220.
[9] 秦正龍,冷 檢,馮長君.密蒙花揮發油成分的定量結構與色譜保留關系研究[J].湖北農業科學,2015,54(3):674-675.
[10] 簡嫦娥,秦正龍,馮長君.木香花揮發油組分的定量結構-保留相關性[J].湖北農業科學,2016,55(11):2882-2884.
[11] FENG C J,YANG W H. Linear QSAR regression models for the prediction of bioconcentration factors of chloroanilines in fish by density functional theory[J].Chinese J Struct Chem,2014,33(6):830-834.
[12] DOLEZAL R,SOUKUP O,MALINAK D,et al. Towards understanding the mechanism of action of antibacterial N-alkyl-3-hydroxypyridinium salts:Biological activities,molecular modeling and QSAR studies[J].European Journal of Medicinal Chemistry,2016,121(4):699-711.
[13] HAWKINS D M,BASAK S C,MILLS D. Assessing model fit by cross-validation[J].J Chem Inf Comput Sci,2003,43(2):579-586.
[14] 劉 東,章文軍,許 祿.手性羥酸和氨基酸類化合物的構效關系研究[J].化學學報,2009,67(2):145-150.
[15] WEI D B,ZHANG A Q,WU C D,et al. Progressive study and robustness test of QSAR model based on quantum chemical parameters for predicting BCF of selected poly-chlorinated organic compounds[J].Chemosphere,2001,44(6):1421-1428.
