銀負載量對網(wǎng)狀TiO2柔性薄膜電極儲鋰性能的影響
2017-07-05 14:55:31張英杰劉嘉銘趙金保黃令李雪
無機化學學報 2017年5期
關(guān)鍵詞:改性
張英杰 劉嘉銘 趙金保 黃令 李雪*,
(1昆明理工大學冶金與能源工程學院,云南省先進電池及材料工程實驗室,昆明650093)
(2廈門大學化學化工學院,固體表面物理化學國家重點實驗室,廈門361005)
銀負載量對網(wǎng)狀TiO2柔性薄膜電極儲鋰性能的影響
張英杰1劉嘉銘1趙金保2黃令2李雪*,1
(1昆明理工大學冶金與能源工程學院,云南省先進電池及材料工程實驗室,昆明650093)
(2廈門大學化學化工學院,固體表面物理化學國家重點實驗室,廈門361005)
采用水熱法結(jié)合銀鏡反應制備出一系列不同Ag負載量(2.2%、4.0%、6.4%,w/w)改性的3D納米網(wǎng)狀結(jié)構(gòu)Ag@TiO2薄膜電極。利用電感耦合等離子體技術(shù)(ICP)、X射線衍射(XRD)、掃描電鏡(SEM)、透射電鏡(TEM)和X射線能譜(EDX)等表征手段測試所合成材料的形貌及成分,實驗結(jié)果表明Ag納米顆粒可以成功沉積在TiO2納米線表面。電化學測試數(shù)據(jù)則表明,4.0%(w/w)負載量的Ag@TiO2相比于未改性和其他負載量的TiO2納米線具有更好的倍率性能和更穩(wěn)定的可逆容量。在50,100,200,400, 800和1 200 mA·g-1的電流密度條件下,該改性電極的放電容量可分別達到261.4,253.7,239.5,216.5,193.1和185.1 mAh·g-1,在200 mA·g-1下循環(huán)80次后容量保持率仍能達到99.8%。
鋰離子電池;柔性薄膜電極;鈦基材料;銀鏡反應
0 引言
鋰離子電池因具有能量密度高、循環(huán)壽命長和無記憶性等優(yōu)點,最近十年呈爆炸式發(fā)展,在人們生活中應用較為廣泛[1-3]。其中TiO2鋰離子電池負極是一種低成本、無毒性和環(huán)境友好的材料,它具備較高的電解液化學兼容性和安全性,被認為是一種石墨碳負極的替代材料[4]。此外,銳鈦礦型TiO2材料中鋰離子的嵌入/脫嵌電壓(>1.0 V vs Li/Li+)明顯高于石墨碳負極的工作電壓,可以有效避免鋰沉積造成的安全隱患[5-7]。得益于銳鈦礦型TiO2材料穩(wěn)定的結(jié)構(gòu),在充放電過程中它體現(xiàn)出優(yōu)秀的容量可逆性和鋰離子快速傳輸行為[8]。這些優(yōu)點說明TiO2負極材料有希望應用在大規(guī)模和長壽命的儲能電池中。
然而,TiO2中的Ti呈+4價,其3d軌道為滿電狀態(tài),沒有可自由移動的電子,導致能帶間隙很寬約為3 eV[9-10],從而嚴重影響TiO2快速轉(zhuǎn)移電子的能力,限制了TiO2在高能量電池領(lǐng)域的應用。目前,提高TiO2材料電化學性能通常采用以下幾種方法:(1)合成納米結(jié)構(gòu)TiO2材料以縮短鋰離子的擴散距離[11-14];(2)進行結(jié)構(gòu)摻雜,產(chǎn)生大量氧空位及三價Ti以提高導電性[15];(3)或與某些導電率高的材料(C, Ag,Au,CNT,PPy等)復合從而降低材料阻抗[16-20]。
在本項研究工作中,我們介紹了一種簡單、無模板合成3D納米網(wǎng)狀結(jié)構(gòu)TiO2薄膜電極材料的方法,并通過傳統(tǒng)銀鏡反應均勻負載上高導電相納米Ag顆粒。最終得到的Ag@TiO2材料具有較大的比表面積和充分的孔洞結(jié)構(gòu),從而提高Li+的快速轉(zhuǎn)移能力和電解液浸潤電極材料的能力。另外,通過銀鏡反應均勻沉積的Ag納米顆粒因具有較高的導電性、分散性及強結(jié)合性使得Ag@TiO2負極材料擁有卓越的倍率性能和循環(huán)性能。
1 實驗部分
1.1 TiO2柔性電極的合成
水熱過程選用一片厚度為0.05 mm的商業(yè)Ti箔(純度99.5%)作為Ti源和基底[21-22],將Ti箔先后放入乙醇和丙酮中超聲洗滌30 min,干燥后的Ti箔豎直放入100 mL內(nèi)襯為聚四氟乙烯高壓釜內(nèi),之后分別加入10 mL乙醇和80 mL濃度為0.5 mol·L-1的NaOH溶液。高壓釜以3℃·min-1的加熱速率升溫至220℃并保溫16 h,再自然冷卻至室溫,即可得到前驅(qū)體。用去離子水清洗數(shù)次后,前驅(qū)體被浸沒在100 mL濃度為0.1 mol·L-1的HCl中6 h,以便將Na+替換為H+[23]。再次用去離子水清洗數(shù)次后,讓前驅(qū)體在室溫下干燥。最后,將已制得的前驅(qū)體放入馬弗爐內(nèi),在空氣氣氛下以3℃·min-1的加熱速率升溫至400℃并保溫4 h。當樣品在室溫下冷卻后,既得TiO2柔性電極。
1.2 Ag@TiO2柔性薄膜電極的制備
Ag納米顆粒通過傳統(tǒng)的銀鏡反應沉積到TiO2電極表面[18,24]。將之前獲得的TiO2電極放入1 L的燒杯底部,分別加入35、70和105 mg的AgNO3及100 mL去離子水。之后在超聲分散下逐滴加入100 mL濃度為5 mmol·L-1的NaOH溶液,待生成棕色沉淀后再逐滴加入10 mol·L-1的濃氨水直至棕色沉淀完全溶解。根據(jù)反應式1和2[25],所得溶液變?yōu)闊o色狀態(tài)。緊接著100 mL濃度為10 mmol·L-1的葡萄糖溶液在強力超聲分散下逐滴滴入上述溶液,并在室溫下反應3 h,發(fā)生反應3[25]。等待反應結(jié)束后,將產(chǎn)物取出并用去離子水和乙醇輪流清洗數(shù)次,60℃真空干燥12 h后即可獲得Ag@TiO2柔性薄膜電極。

1.3 樣品的表征
采用Rigaku MiniFlex600 X′pert型X射線衍射儀(XRD,Cu靶,λ=0.154 05 nm)進行物相分析,用HITACHI S-4800型場發(fā)射掃描電子顯微鏡(FESEM)和JEM-2100型高分辨率透射電鏡(HRTEM)觀察樣品的形貌和結(jié)構(gòu),利用HITACHI S-4800型掃描電鏡附帶的X射線能譜分析儀(EDX)來分析元素含量及分布情況,通過電感耦合等離子體技術(shù)(ICP)測定Ag@TiO2材料中的Ti,Ag元素含量。
1.4 電池組裝和性能表征
TiO2和Ag@TiO2柔性電極作為研究電極,以金屬鋰片為對電極,Celgard 2400聚丙烯微孔膜為隔膜,以1 mol·L-1LiPF6的EC+DMC+DEC(1∶1∶1,V/V)為電解液,在氬氣氛手套箱里組裝成CR2016型扣式電池。在LAND-V34電池性能測試系統(tǒng)上進行恒電流充放電實驗,電壓范圍為1.0~3.0 V。在MetrohmAutolabPGSTAT302N型電化學工作站上進行循環(huán)伏安(CVs)和交流阻抗(EIS)測試。在SX1934 (SZ-82)型四探針電阻測試儀上測試TiO2和Ag@TiO2電極的薄層電阻。
2 結(jié)果與討論
通過ICP分析改性產(chǎn)物中銀的質(zhì)量分數(shù)(見表1),將改性后的材料分別記為Ag2.2@TiO2、Ag4.0@TiO2和Ag6.4@TiO2。銀改性后的TiO2納米線XRD圖如圖1所示。由圖1可知,Ag@TiO2納米線衍射峰的2θ=25.4°,37.8°,48.1°,62.8°和76.2°與銳鈦礦型TiO2(JCPDS No.89-4921)[18,26]相吻合,而2θ= 40.2°,53.0°和70.6°與金屬Ti(JCPDS No.89-2762)相吻合,Ag(JCPDS No.87-0717)的特征峰則在38.1°, 44.3°,64.4°和77.4°出現(xiàn),并隨著銀含量的增加而逐漸加強。這說明通過銀鏡反應,Ag成功負載在TiO2納米線上并且沒有對其晶體結(jié)構(gòu)造成負面的影響。

表1 ICP測試所得Ag@TiO2納米材料中元素Ag的含量Table 1Elemental contents(in weight percent)of Ag in the Ag-modified TiO2samples obtained via ICP testing method

圖1 不同Ag含量的Ag@TiO2納米材料XRD圖Fig.1XRD patterns of Ag@TiO2nanomaterials
圖2 為TiO2和Ag改性后TiO2樣品的SEM圖片以及Ag4.0@TiO2樣品的EDX元素分布圖譜。如圖2a和b可知,所得樣品由大量無序分布、大小均勻的1D納米線組成,最終呈現(xiàn)3D納米網(wǎng)狀結(jié)構(gòu)。薄膜電極這種特殊的3D納米網(wǎng)狀結(jié)構(gòu)優(yōu)勢明顯,不僅可以縮短Li+離子的擴散途徑,而且大的比表面積有利于電解液的浸潤和提高Li+離子的傳輸速度。從圖2c~e可知,改性后的TiO2仍保持3D納米網(wǎng)狀結(jié)構(gòu),納米Ag顆粒粒徑在10~30 nm范圍內(nèi),且均勻地負載在TiO2納米線表面。我們發(fā)現(xiàn)隨著含量的增加,Ag納米顆粒逐漸增大,Ag顆粒的分散性也隨Ag含量的增加而變化:其中含Ag量為2.2%(w/w)時,Ag顆粒較小,因此具有較大的表面活性能,這個較高的表面活性能不但使Ag顆粒易于團聚,同時也導致負載Ag粒子的TiO2納米線易于產(chǎn)生團聚。而Ag負載量增加到6.4%(w/w)時Ag粒子粒徑發(fā)生了急速增長,導致它的分散性較差。整體說來,含Ag量適中的Ag4.0@TiO2電極具有最佳的Ag粒子分散性,同時TiO2納米線也不容易發(fā)生團聚。因此,我們認為Ag4.0@TiO2電極在不犧牲TiO2納米網(wǎng)多孔結(jié)構(gòu)的情況下,將獲得最佳的導電性能。高分散的Ag粒子與TiO2三維多孔的納米網(wǎng)狀結(jié)構(gòu)不但可以提高材料的導電性,也利于電解液的浸潤與離子、電子的傳導,最終可以整體提高TiO2復合電極的電化學性能。在之后的電化學測試中,Ag4.0@TiO2樣品的電化學性能最佳,因此后續(xù)實驗著重對它進行了結(jié)構(gòu)表征及電化學性能測試。其中EDX測試(見圖2f)測試結(jié)果(見表2)與ICP分析結(jié)果一致,這進一步證明Ag已有效地均勻覆蓋在TiO2柔性電極表面。
圖3是Ag納米顆粒在Ag4.0@TiO2納米線中分布的TEM圖。從圖3a可知,Ag納米顆粒不僅負載在單一的納米線上,也成功負載在納米線的連接處,從而形成完整均勻的導電網(wǎng)絡。這些Ag納米顆粒的尺寸約為10~30 nm,與SEM圖結(jié)果一致。圖3c和d分別為Ag4.0@TiO2納米線的高分辨透射電鏡(HRTEM)圖和選區(qū)電子衍射(SAED)圖,由圖可知該納米線是高度結(jié)晶的,此外,采用Gatan Digital Micrograph軟件對材料的晶格條紋分析發(fā)現(xiàn),d= 0.352 nm與TiO2的(101)晶面間距(d(101)=0.351 nm)相符合,進一步表明該TiO2納米線的結(jié)晶相為銳鈦礦相。從材料的SAED圖可知,該納米線為單晶結(jié)構(gòu),并且對SAED圖中的透射斑分析發(fā)現(xiàn),TiO2納米線所產(chǎn)生的晶面有(101)、(200),這些均與XRD結(jié)果相吻合。納米線的生長方向與(101)面的夾角約為71.5°,這一角度值與文獻報道的銳鈦礦型TiO2(101)面和[100]方向所呈的夾角相一致[20,27-28],這也表明了該水熱反應法制備得到的TiO2納米線的生長方向為[100]方向。

圖2 TiO2(a)和Ag4.0@TiO2(b)的放大SEM圖及對應的EDX圖譜(f),Ag2.2@TiO2(c)、Ag4.0@TiO2(d)和Ag6.4@TiO2(e)的SEM圖Fig.2SEM images of(a)TiO2,(b)Ag4.0@TiO2,magnified SEM images of(c)Ag2.2@TiO2,(d)Ag4.0@TiO2and(e)Ag6.4@TiO2; (f)Corresponding surface EDX spectrum of(b)
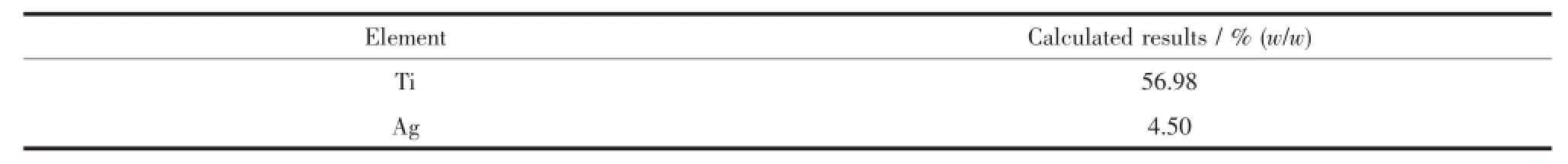
表2 EDX測試所得Ag4.0@TiO2樣品中元素Ag和Ti的含量Table 2Elemental contents(in weight percent)of Ag and Ti in the Ag4.0@TiO2samples obtained via EDX analysis

圖3 Ag4.0@TiO2的TEM圖(a,b)、HRTEM圖(c)和(b)圖對應的SAED圖(d)Fig.3Bright-filed TEM images(a,b)Ag4.0@TiO2;(c)corresponding HRTEM images and(d)SAED pattern from selected area in(b)
圖4a是TiO2和Ag@TiO2電極在電流密度200 mA·g-1下的放電容量循環(huán)穩(wěn)定性圖,TiO2電極在第二圈的放電容量為262.3 mAh·g-1,80次循環(huán)后容量為157.8 mAh·g-1,容量保持率為60.3%。雖然Ag4.0@TiO2和Ag6.4@TiO2的初始容量略低于TiO2,但所有Ag@TiO2電極都擁有更高的容量保持率。特別是Ag4.0@TiO2電極在80次循環(huán)后容量保持率高達99.8%,放電容量仍能達到225.6 mAh·g-1,這說明Ag4.0@TiO2材料具有出眾的循環(huán)穩(wěn)定性。圖4b的庫倫效率對比圖中,我們發(fā)現(xiàn)Ag顆粒的負載都在一定程度上提高了材料的首次庫倫效率,這說明Ag的負載提高了材料的導電性能,從而提高了首次充放電過程的可逆性。從庫倫效率的穩(wěn)定性分析,我們發(fā)現(xiàn)隨著Ag負載量的增加,材料的庫倫效率在不同程度上趨于穩(wěn)定,但是當Ag負載量高達6.4%時,庫倫效率的穩(wěn)定性改善的不夠明顯。我們認為這是因為,Ag沉積過多,會導致更多的二氧化鈦活性面被遮擋而無法進行更好的儲鋰反應,所以,雖然導電性能得到穩(wěn)定的提高,其電化學反應的活性面卻減少了。圖4c對比了TiO2和Ag@TiO2電極在不同電流密度下的倍率性能。Ag2.2@TiO2和Ag4.0@TiO2電極在所有電流密度下都展示了明顯高于TiO2的可逆容量,并且這兩種電極在低電流密度時的可逆容量差距較小,然而在更高電流密度如1 200 mA·g-1時,Ag4.0@TiO2電極的放電容量可達193.6 mAh· g-1,明顯高于Ag2.2@TiO2(179.8 mAh·g-1)與Ag6.4@ TiO2電極(155.9 mAh·g-1)。總而言之,Ag4.0@TiO2電極的電化學性能最為優(yōu)秀。
圖5是TiO2和Ag4.0@TiO2電極在電流密度為200 mA·g-1下的充放電曲線。從圖中可以觀察到充放電平臺大約在2.0和1.75 V(vs Li+/Li),TiO2電極的放電曲線可以被分為3個連續(xù)的區(qū)域[10,22,29],區(qū)域1是從開路電壓迅速降低至放電平臺(約1.75 V)處的電壓區(qū)間段,該區(qū)間段是由于少量Li+嵌入至I41/ amd四方晶系銳鈦礦相TiO2晶格中,生成LixTiO2(I41/amd tetragonal anatase,x<0.5)的固溶相反應過程[29-30]。區(qū)域2對應的是1.75 V(vs Li/Li+)的放電平臺,該區(qū)域?qū)氖秦氫囅郘ixTiO2(x<0.5)和富鋰相Li0.5TiO2(orthorhombic)之間的兩相反應過程[14,29-30]。區(qū)域3是從放電平臺至截止電壓1.0 V處的電壓平緩降低區(qū)間段,該區(qū)間段主要是由于納米級TiO2材料的表面發(fā)生贗電容嵌鋰反應,使得Li+在材料表面進一步嵌入,生成Li0.5+xTiO2(0<x≤0.5)所引起的[17],此類現(xiàn)象在其他納米TiO2材料中也有報道[22,31-32]。從圖5b可知,改性后和未改性的TiO2相比,充放電容量沒有較大區(qū)別,充放電平臺也沒有差別,說明Ag納米顆粒不存在電化學活性,僅扮演提高TiO2導電性的角色并不能增加其容量。

圖4 TiO2和Ag@TiO2電極(a)放電容量循環(huán)圖,電流密度為200 mA·g-1,(b)庫倫效率圖,(c)倍率性能圖,電流密度50、100、200、400、800和1 200 mA·g-1Fig.4(a)Cycle performances for TiO2and Ag@TiO2electrodes at a current density of 200 mA·g-1;(b)Related coulomb efficiency; (c)Rate capabilities of TiO2and Ag@TiO2electrodes at different current densities

圖5 (a)TiO2和(b)Ag4.0@TiO2電極的充放電曲線圖Fig.5Charge and discharge curves of(a)TiO2and(b)Ag4.0@TiO2electrodes at a current density of 200 mA·g-1
圖6 給出的是TiO2與Ag4.0@TiO2電極在掃描速度0.1、2 mV·s-1時的循環(huán)伏安曲線。圖中循環(huán)伏安曲線所圍成的面積代表法拉第和非法拉第反應過程所產(chǎn)生的總的存儲電荷量。從圖中CV曲線可以看到,兩種電極材料在2.0 V/1.75 V和1.65 V/1.55 V處都出現(xiàn)了氧化/還原電對峰。根據(jù)Kubiaka和Armstrong等的研究報道[32-33]認為,位于2.0 V/1.75 V處的氧化/還原峰(簡寫為A峰)對應的是Li+在TiO2晶格中的固溶相嵌入和脫出反應過程,而位于1.65 V/1.55 V處的氧化/還原峰(簡寫為S峰)對應的是Li+在TiO2表面的贗電容嵌入和脫出反應過程。其中,S峰對應的贗電容嵌脫鋰容量與銳鈦礦TiO2材料的比表面積成正比關(guān)系,而與材料的尺寸大小成反比關(guān)系。另外,一些研究者認為CV中出現(xiàn)的S峰屬于單斜晶系TiO2(B)的特征峰[34-35],然而通過XRD的分析結(jié)果表明TiO2材料并無TiO2(B)相的存在,我們認為出現(xiàn)這一現(xiàn)象的原因則可能是由于更加開放的納米級3D網(wǎng)狀結(jié)構(gòu)和具有較大比表面積的一維納米線所共同導致的[36]。相比未于改性的電極材料,改性電極Ag4.0@TiO2在更快的掃描速度下具有更多的存儲電荷量,說明其電化學可逆性更好。

圖6 (a)TiO2在0.1 mV·s-1下的循環(huán)伏安圖,(b)Ag4.0@TiO2在0.1 mV·s-1下的循環(huán)伏安圖,(c)TiO2在2 mV·s-1下的循環(huán)伏安圖,(d)Ag4.0@TiO2在2 mV·s-1下的循環(huán)伏安圖Fig.6CV measurements of(a)TiO2and(b)Ag4.0@TiO2at a scan rate of 0.1 mV·s-1;CV measurements of(c)TiO2and (d)Ag4.0@TiO2at 2 mV·s-1
通過交流阻抗測試和四點探針測試,可以進一步了解材料的電化學性能。交流阻抗被認為是了解電極動力學過程最有效的測試之一[37-38],圖7是TiO2和Ag4.0@TiO2電極的交流阻抗測試圖、擬合后的數(shù)據(jù)圖以及等效電路圖,測試電位為開路電壓約為3.0 V(vs Li/Li+)。圖中每條曲線都由高頻區(qū)的半圓和低頻區(qū)的直線部分組成,高頻半圓與電荷轉(zhuǎn)移阻抗相關(guān),而低頻直線區(qū)則與長程鋰離子擴散的Warburg阻抗相關(guān)[32]。圖7c的等效電路擬合誤差小于6%,符合分析要求,在此等效電路中,Rs代表電池中的歐姆阻抗,包括電極、隔膜、導體、活性物質(zhì)等的電阻,Rct代表活性物質(zhì)表面的電荷轉(zhuǎn)移阻抗,CPE代表雙電層電容和鈍化膜電容,Ws代表Warburg阻抗。表3是等效電路擬合的參數(shù)數(shù)據(jù),Ag4.0@TiO2電極的阻抗明顯低于未改性的TiO2電極,這說明采用Ag納米顆粒負載在TiO2材料表面能夠切實有效地提高導電率。表4是四點探針的薄膜電阻結(jié)果,Ag4.0@TiO2電極的薄膜電阻(168.92 Ω·sq-1)顯著低于TiO2電極電阻(2.5750×105Ω·sq-1),進一步驗證Ag納米顆粒改性有利于提高TiO2電極的導電性。

圖7 (a)TiO2和(b)Ag4.0@TiO2電極交流阻抗測試圖譜和(c)等效電路圖,測試電位為開路電壓約3.0 V(vs Li/Li+)Fig.7AC impedance measurements of(a)TiO2and(b)Ag4.0@TiO2electrode at open-circuit potential of~3.0 V(vs Li/Li+); (c)Related equivalent electric circuit

表3 TiO2和4.0Ag@TiO2電極在開路電壓約為3.0 V(vs Li/Li+)的交流阻抗參數(shù)Table 3AC impedance parameters of TiO2and Ag4.0@TiO2electrodesopen-circuit potential of~3.0 V(vs Li/Li+)

表4 TiO2和4.0Ag@TiO2電極通過四探針測試所得薄膜電阻結(jié)果Table 4Sheet resistances of TiO2and Ag4.0@TiO2electrodes measured by the four-point probe testing method
3 結(jié)論
我們通過水熱法結(jié)合傳統(tǒng)銀鏡反應制備了Ag負載改性的TiO2薄膜電極材料。在電化學測試中,Ag@TiO2薄膜電極作為鋰電池負極展示出高倍率和高穩(wěn)定性能。在倍率測試中電流密度1 200 mA·g-1條件下,Ag4.0@TiO2電極的放電容量為194.2 mAh· g-1,容量保持率為71.5%。不僅如此,在較高電流密度200 mA·g-1下80次循環(huán)后容量基本沒有衰減,容量保持率高達99.8%,明顯優(yōu)于其他Ag負載量的TiO2電極。Ag4.0@TiO2電極優(yōu)良的電化學性能來源于獨特的3D納米網(wǎng)狀結(jié)構(gòu)和Ag改性帶來的更快的離子轉(zhuǎn)移速度和更高的導電率。此改性方法是解決TiO2電極低導電率和倍率性能問題的有效途徑,我們相信它具有更廣闊地應用空間,Ag@TiO2是一種有前景的高倍率鋰離子電池負極材料。
[1]ZHANG Yu(張鈺),SU Zhi(粟智),PAN Hui(潘會).Chinese J.Inorg.Chem.(無機化學學報),2015,31(9):1827-1830
[2]HU Guo-Gong(胡國榮),LU Wei(盧葦),LIANG Long-Wei (梁龍偉),et al.Chinese J.Inorg.Chem.(無機化學學報), 2015,31(1):159-165
[3]XIA Shu-Biao(夏書標),ZHANG Ying-Jie(張英杰),DONG Peng(董鵬),et al.Chinese J.Inorg.Chem.(無機化學學報),2014,30(3):529-535
[4]Cho M Y,Park K B,Roh K C.Electron.Mater.Lett.,2013,9 (6):809-812
[5]Lou X W,Archer L A.Adv.Mater.,2008,20(10):1853-1858
[6]LIU Mei-Pin(劉美玭),HU Yu-Xiang(胡宇翔),DU Hong-Bin (杜紅賓).Chinese J.Inorg.Chem.(無機化學學報),2015,31 (12):2425-2431
[7]Wang D,Choi D,Li J,et al.ACS nano,2009,3(4):907-914
[8]Yang Y,Ji X,Jing M,et al.J.Mater.Chem.A,2015,3(10): 5648-5655
[9]Wang H,Yan Y,Chen G.J.Mater.Chem.A,2015,3(19): 10275-10283
[10]Kim S K,Son M K,Park S,et al.Electron.Mater.Lett., 2014,10(1):229-234
[11]Kavan L,Gr?tzel M,Rathousky J,et al.J.Electrochem. Soc.,1996,143(2):394-400
[12]Sudant G,Baudrin E,Larcher D,et al.J.Mater.Chem., 2005,15(12):1263-1269
[13]Moriguchi I,Hidaka R,Yamada H,et al.Adv.Mater., 2006,18(1):69-73
[14]Pan X,Yang M Q,Fu X,et al.Nanoscale,2013,5(9):3601-3614
[15]Moriguchi I,Hidaka R,Yamada H,et al.Adv.Mater., 2006,18(1):69-73
[16]Nam S H,Shim H S,Kim Y S,et al.ACS Appl.Mater. Interfaces,2010,2(7):2046-2052
[17]Li N,Zhou G,Fang R,et al.Nanoscale,2013,5(17):7780-7784
[18]He B L,Dong B,Li H L.Electrochem.Commun.,2007,9(3): 425-430
[19]Tang W,Gao X,Zhu Y,et al.J.Mater.Chem.A,2012,22 (38):20143-20145
[20]Liu Y,Zhang B H,Xiao S Y,et al.Electrochim.Acta, 2014,116(2):512-517
[21]Peng X,Chen A.Adv.Funct.Mater.,2006,16(10):1355-1362
[22]Liao J Y,Lei B X,Chen H Y,et al.Energy Environ.Sci., 2012,5(2):5750-5757
[23]Kasuga T,Hiramatsu M,Hoson A,et al.Adv.Mater., 1999,11(15):1307-1311
[24]Rahman M M,Wang J Z,Wexler D,et al.J.Solid State Electrochem.,2010,14(4):571-578
[25]Huang S,Wen Z,Zhang J,et al.Electrochim.Acta,2007, 52(11):3704-3708
[26]Gentili V,Brutti S,Hardwick L J,et al.Chem.Mater.,2012, 24(22):4468-4476
[27]Liu B,Boercker J E,Aydil E S.Nanotechnology,2008,19 (50):505604-505610
[28]Boercker J E,Enache-Pommer E,Aydil E S.Nanotechnology, 2008,19(9):095604-095613
[29]RenY,HardwickLJ,BrucePG.Angew.Chem.Int.Ed.,2010, 122(14):2624-2628
[30]Shin J Y,Samuelis D,Maier J.Adv.Funct.Mater.,2011,21 (18):3464-3472
[31]Kim S W,Han T H,Kim J,et al.ACS Nano,2009,3(5): 1085-1090
[32]Wang J,Polleux J,Lim J,et al.J.Phys.Chem.C,2007,111 (40):14925-14931
[33]Armstrong G,Armstrong A R,Canales J,et al.Electrochem. Solid.ST.,2006,9(3):A139-A143
[34]Zukalova M,Kalbac M,Kavan L,et al.Chem.Mater.,2005, 17(5):1248-1255
[35]Prochzka J,Kavan L,Zukalova M,et al.Chem.Mater.,2009, 21(8):1457-1464
[36]Laskova B,Zukalova M,Zukal A,et al.J.Power Sources, 2014,246(3):103-109
[37]Rahman M M,Wang J Z,Wexler D,et al.J.Solid State Electrochem.,2010,14(4):571-578
[38]Li X,Lin H,Cui W,et al.ACS Appl.Mater.Interfaces, 2014,6(10):7895-7901
Effect of Loading Content of Silver on Lithium Storage for TiO2Net-Work Flexible Film Electrode
ZHANG Ying-Jie1LIU Jia-Ming1ZHAO Jin-Bao2HUANG Ling2LI Xue*,1
(1Engineering Laboratory of Advanced Battery and Materials of Yunnan Province,Faculty of Metallurgical and Energy Engineering, Kunming University of Science and Technology,Kunming 650093,China)
(2State Key Laboratory of Physical Chemistry of Solid Surfaces,Department of Chemistry,College of Chemistry and Chemical Engineering,Xiamen University,Xiamen,Fujian 361005,China)
A series of silver-modified TiO2flexible film electrodes with hierarchical 3D nano-network structure are facilely synthesized using hydrothermal method followed via the traditional silver mirror reaction.Successful deposition of Ag nanoparticles on to the TiO2nanowires surface is confirmed by inductively coupled plasma(ICP) technique,X-ray diffraction(XRD),scanning electron microscope(SEM)with energy-dispersive X-ray analysis (EDX)and transmission electron microscopy(TEM).The electrochemical properties of the Ag@TiO2nanowires were researched in this work.Our results show that the Ag@TiO2nanowires with 4.0%(w/w)silver exhibits a better rate performance and more excellent reversibility than pristine one.At different current densities of 50, 100,200,400,800 and 1 200 mA·g-1,the discharge capacities of the Ag@TiO2electrodes are 261.4,253.7, 239.5,216.5,193.1 and 185.1 mAh·g-1,respectively.After 80 cycles at 200 mA·g-1,its discharge capacity retention is 99.8%indicating Ag@TiO2electrodes are potentially useful for LIBs.
lithium ion battery;flexible film electrodes;Ti-based materials;silver mirror reaction
O646.6
A
1001-4861(2017)05-0809-08
10.11862/CJIC.2017.087
2016-10-24。收修改稿日期:2017-03-13。
國家自然科學基金(No.51604132)資助項目。
*通信聯(lián)系人。E-mail:4386160734@qq.com
猜你喜歡
紡織科學研究(2020年1期)2020-05-21 00:31:06
中國塑料(2016年12期)2016-06-15 20:30:07
中國塑料(2016年2期)2016-06-15 20:30:00
中國塑料(2016年2期)2016-06-15 20:29:59
中國塑料(2016年5期)2016-04-16 05:25:36
廣西林業(yè)科學(2016年3期)2016-03-16 05:43:30
中國塑料(2015年3期)2015-11-27 03:41:38
中國塑料(2015年11期)2015-10-14 01:14:14
中國塑料(2015年9期)2015-10-14 01:12:17
中國塑料(2015年4期)2015-10-14 01:09:19

- 無機化學學報的其它文章
- Three Coordination Complexes Based on an Asymmetric Triazole Derivative Ligand: Syntheses,Structures and Photocatalytic Properties
- Two Tetrapodal Schiff Bases Acting as Colorimetric Sensors for Iron in Environmental Water Samples
- Syntheses,Characterization and Fluorescence Properties Analysis of Two Lanthanide Coordination Polymers Based on Axial Chirality Ligand 2,2′-Dinitro-4,4′-biphenyldicarboxylic Acid
- 磁性UiO-66復合材料的合成及其對水體中硝基酚有機分子的吸附性能
- cis-[(trans-1,2-雙(氨甲基)環(huán)丁烷) (3-羥基-1,1-環(huán)丁烷二羧酸根)合鉑(Ⅱ)]的合成和抗癌活性
- 兩個具有Sn4O4梯狀結(jié)構(gòu)二丁基錫羧酸酯的微波溶劑熱合成、結(jié)構(gòu)和體外抗癌活性